Administración Nacional de Medicamentos, Alimentos y Tecnología Médica
PRODUCTOS PARA DIAGNOSTICO
Disposición 2009/2007
Establécense las pautas, documentación y requisitos a presentar para solicitar la autorización de los medicamentos destinados a ser administrados a seres humanos con fines diagnósticos o de monitoreo denominados Productos para Diagnóstico de uso "in vivo".
Bs. As., 9/4/2007
VISTO el Expediente Nº 1-47-1110-4309-04-8 del Registro de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica, y
CONSIDERANDO:
Que el Decreto Nº 150/92 define como medicamento a: "Toda preparación o producto farmacéutico empleado para la prevención, diagnóstico y/o tratamiento de una enfermedad o estado patológico, o para modificar sistemas fisiológicos en beneficio de la persona a quien se le administra".
Que la Resolución (ex M.S. y A.S.) Nº 102/98 define los Productos para Diagnóstico de Uso "in vivo" como "productos administrados a seres humanos con el propósito de contribuir al diagnóstico de una enfermedad u otras condiciones, incluida la evaluación del estado de salud, con la finalidad de curar, tratar o prevenir una enfermedad o sus secuelas, cuya efectividad diagnóstica se encuentre comprobada y que en las dosis o concentraciones administradas no posean efecto terapéutico alguno".
Que los Productos Radio Farmacéuticos destinados al diagnóstico por imágenes se encuentran comprendidos en la definición de Productos y/o Reactivos para Diagnóstico de uso "in vivo" mencionada en el considerando anterior.
Que los productos mencionados destinados a ser administrados a seres humanos con fines diagnósticos o de monitoreo clínico deben reunir requisitos de seguridad y eficacia específicos para los fines propuestos, debiendo estar inscriptos en el Registro de Medicamentos de esta Administración Nacional.
Que en cumplimiento de lo dispuesto por el artículo 4º de la Resolución Ex MS y AS Nº 102/98, aplicable en materia de Productos para Diagnóstico de uso "in vivo", corresponde a esta Administración Nacional de Medicamentos, Alimentos y Tecnología Médica dictar las normas reglamentarias atinentes al registro, fiscalización y control de tales productos.
Que por lo tanto resulta necesario establecer las pautas, documentación y requisitos a presentar para solicitar la autorización de los medicamentos destinados a ser administrados a seres humanos con fines diagnósticos o de monitoreo denominados Productos para Diagnóstico de uso "in vivo".
Que la información y documentación presentadas deben permitir la evaluación de la utilidad diagnóstica declarada del producto, así como su calidad, eficacia y seguridad diagnóstica.
Que los productos utilizados para el diagnóstico por imágenes, en especial las Preparaciones Radiofarmacéuticas administradas a los seres humanos con fines diagnósticos o de monitoreo, deben reunir requisitos de seguridad y eficacia específicos para los fines propuestos.
Que el Instituto Nacional de Medicamentos y la Dirección de Asuntos Jurídicos han tomado la intervención de su competencia.
Que se actúa en virtud de las facultades conferidas por el Decreto Nº 1490/92, el Decreto Nº 197/02 y la Resolución ex M.S. y A.S. Nº 102/98.
Por ello,
EL INTERVENTOR NACIONAL DE LA ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS
Y TECNOLOGIA MEDICA
DISPONE:
Artículo 1º — A los fines de la presente Disposición se consideran Productos para Diagnóstico por Imágenes a aquellos medicamentos que administrados a seres humanos con fines de diagnóstico o monitoreo, son utilizados con técnicas por imágenes tales como la tomografía computada (CT), ultrasonografía, resonancia magnética nuclear, centellografía u otros, quedando clasificados en dos categorías generales:
• Medios de contraste: Medicamentos que comprenden, pero no se limitan, a: compuestos iodados utilizados en radiografía y tomografía computada, iones metálicos paramagnéticos tales como iones de gadolinio, hierro y manganeso unidos a diversas moléculas y utilizados en Resonancia Magnética nuclear, productos utilizados en ultrasonografía diagnóstica.
• Preparaciones Radiofarmacéuticas para Diagnóstico de uso "in vivo": Medicamentos que comprenden productos radiactivos que contienen un radionucleído, pudiendo estar éste unido a un ligando o carrier, y que son utilizados en imágenes planares, tomografía computarizada por emisión de fotón simple (SPECT), tomografía de emisión de positrones (PET) u otras técnicas diagnósticas. A los efectos previstos en esta Disposición, se consideran Preparaciones Radiofarmacéuticas para Diagnóstico de uso "in vivo" o Agentes para Radiodiagnóstico de uso "in vivo" a los productos destinados a ser usados en el diagnóstico o monitoreo de una enfermedad o manifestación de una enfermedad en seres humanos, que exhiben desintegración espontánea de un nucleído inestable con la emisión de partículas nucleares o fotones, estando comprendidos los juegos de reactivos no radiactivos y generadores de nucleídos utilizados en la preparación de estos productos.
Art. 2º — La presente Disposición será de aplicación al registro de los Medicamentos denominados Preparaciones Radiofarmacéuticas para Diagnóstico de uso "in vivo". La presentación de solicitudes de autorización de Preparaciones Radiofarmacéuticas destinados al diagnóstico de uso "in vivo" deberá incluir en forma expresa las indicaciones o destino de uso de los mismos.
Art. 3º — A los fines del Artículo 2º de la presente Disposición, las Preparaciones Radiofarmacéuticas para Diagnóstico de uso "in vivo" quedarán clasificadas en las siguientes categorías según indicaciones de uso:
Categoría 1- Delineación de Estructura: Productos destinados a la localización de estructuras anatómicas normales o a la distinción entre estructuras anatómicas normales y anormales.
Categoría 2- Evaluación del Estado Funcional, Fisiológico o Bioquímico: Productos destinados a la evaluación de procesos funcionales, fisiológicos o bioquímicos normales cuando una alteración en los mismos sean comunes a diversas patologías o condiciones, no teniendo indicaciones diagnósticas para una patología o condición en particular.
Categoría 3- Establecimiento o Detección de una Enfermedad o Patología: Productos utilizados como ayuda en la detección, localización o caracterización de una enfermedad o estado patológico específico.
Categoría 4- Monitoreo diagnóstico o seguimiento terapéutico de un paciente: Productos que mediante la obtención de imágenes proveen información que permite el monitoreo diagnóstico o la toma de decisiones apropiadas para el seguimiento terapéutico del paciente.
Las indicaciones de uso establecidas para un mismo producto podrán estar comprendidas en más de una de las categorías antes establecidas. Cuando la indicación de uso sea diferente a las contempladas en la clasificación antes presentada, el solicitante deberá señalar la indicación propuesta y los métodos utilizados para demostrar su eficacia.
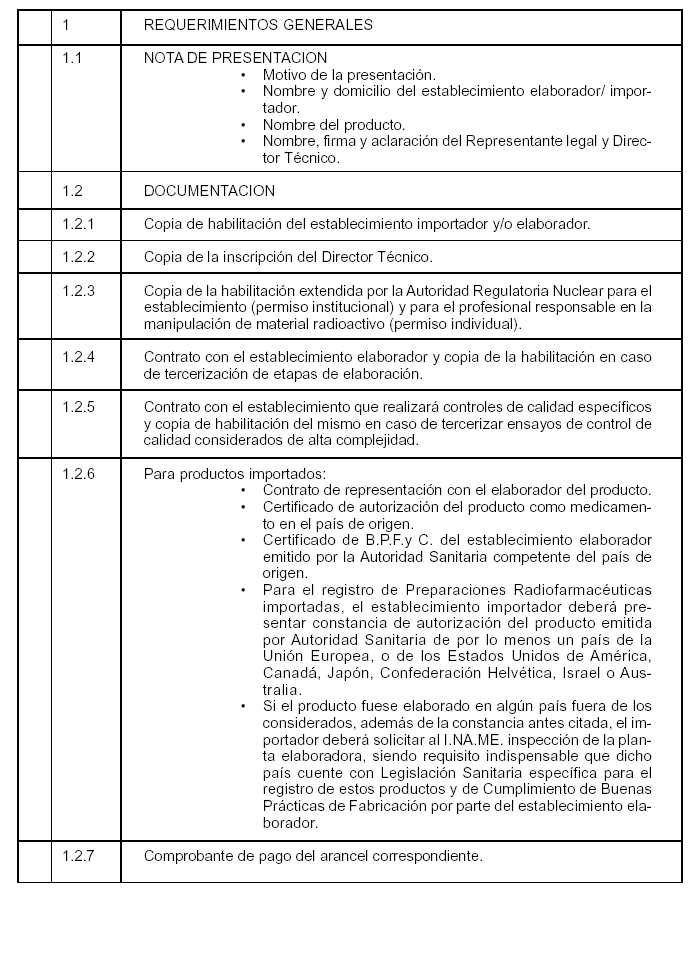
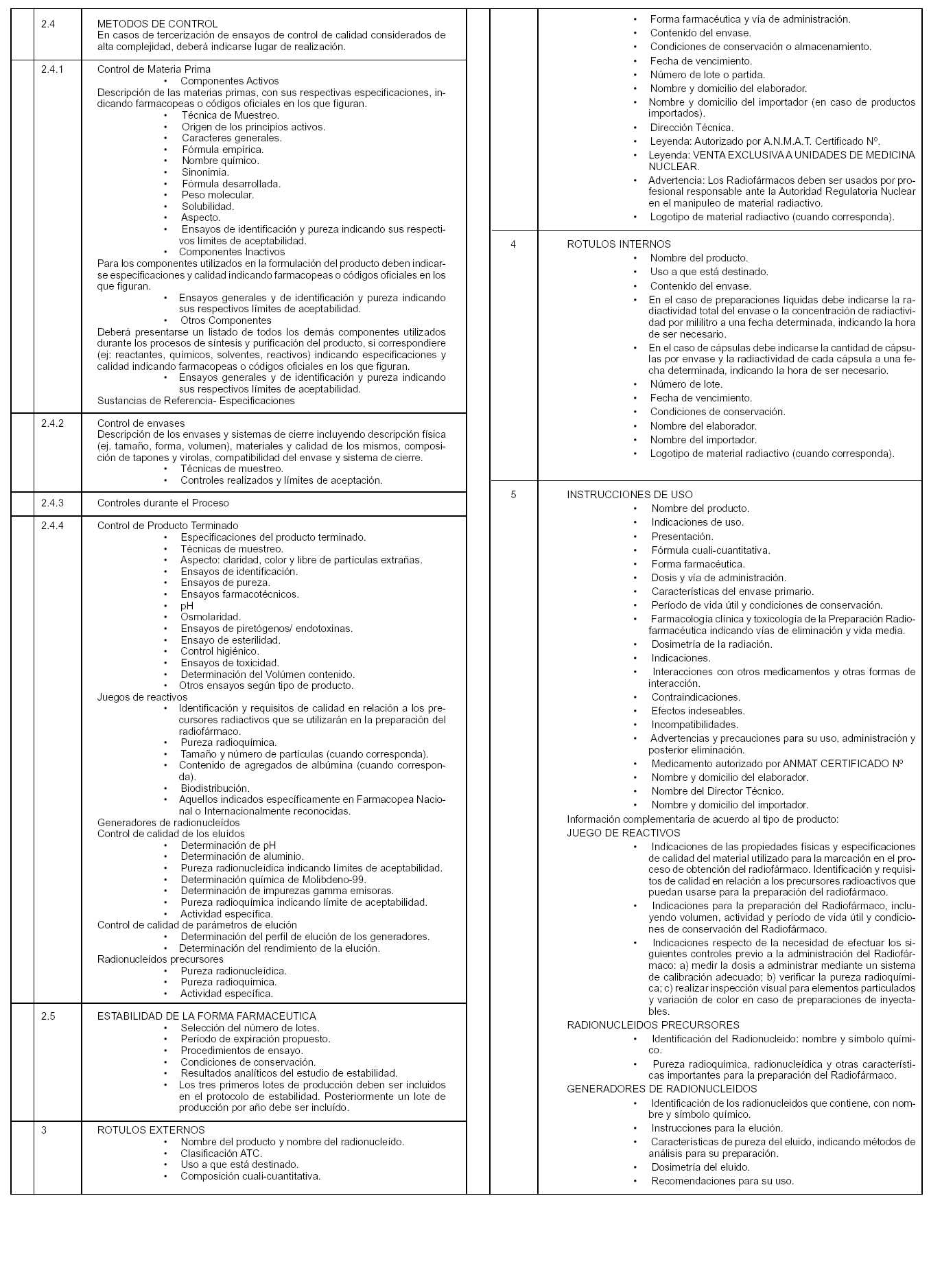
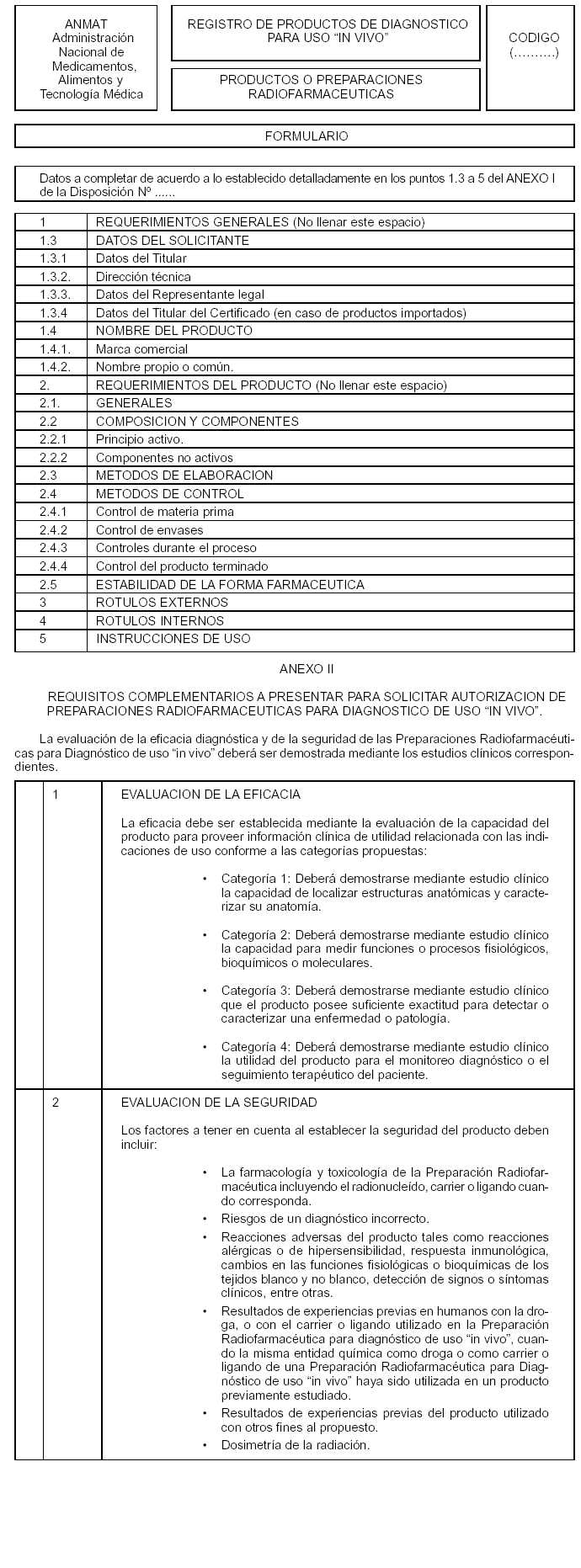
Art. 4º — A fin de solicitar la autorización de una nueva Preparación Radiofarmacéutica para Diagnóstico de uso "in vivo", el establecimiento elaborador y/o importador deberá en todos los casos presentar la documentación e información que consta en el ANEXO I de la presente Disposición y que forma parte integrante de la misma.
Art. 5º — Para el registro de Preparaciones Radiofarmacéuticas importadas, el establecimiento importador deberá presentar además de la documentación e información que consta en el ANEXO I citado en el artículo 4º, constancia de autorización del producto emitida por Autoridad Sanitaria de por lo menos un país de la Unión Europea, USA, Canadá, Japón, Confederación Helvética, Israel o Australia.
Si el producto fuese elaborado en algún país fuera de los considerados, además de la constancia antes citada, el importador deberá solicitar al I.NA.ME. inspección de la planta elaboradora, siendo requisito indispensable que dicho país cuente con legislación sanitaria específica para el registro de estos productos y de Cumplimiento de Buenas Prácticas de Fabricación por parte del establecimiento elaborador.
Art. 6º — La información sobre la eficacia y seguridad de la Preparación Radiofarmacéutica para Diagnóstico de uso "in vivo" deberá ser presentada de conformidad con lo establecido en el ANEXO II de la presente Disposición y que forma parte integrante de la misma.
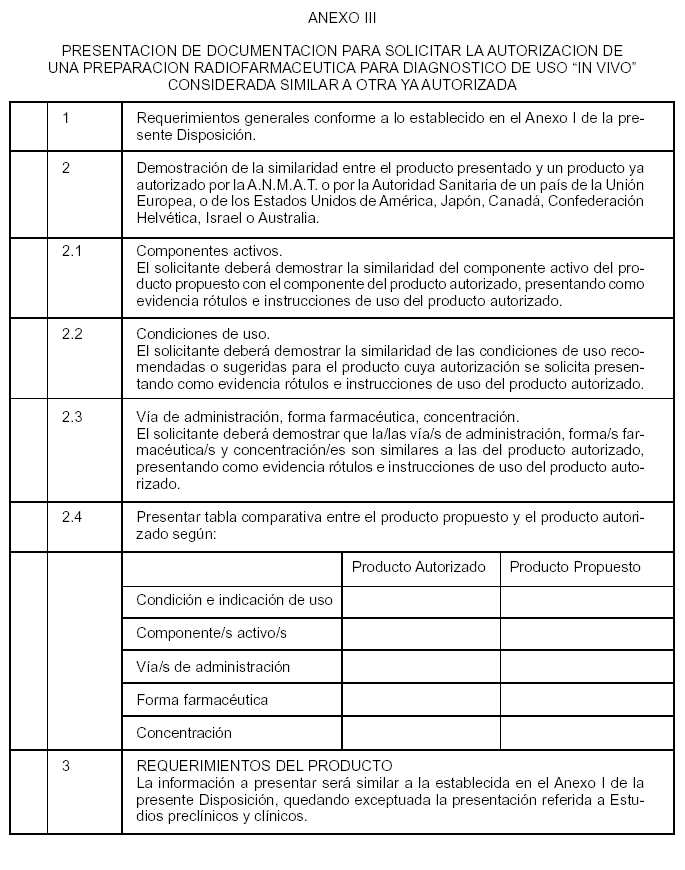
Art. 7º — Para solicitar la autorización de una Preparación Radiofarmacéutica para Diagnóstico de uso "in vivo" considerada similar a otra ya autorizada, se deberá presentar la documentación e información que consta en el ANEXO III de la presente Disposición y que forma parte integrante de la misma.
Art. 8º — En aquellos casos en los que se solicite autorización de una Preparación Radiofarmacéutica para Diagnóstico de uso "in vivo" considerada similar a otra ya autorizada pero que exista una modificación de excipientes o componentes no activos, deberá demostrarse que dicha modificación no altera la seguridad del producto ni la cantidad y/o concentración del principio activo en el producto terminado. La información a presentar será la siguiente: a) Similaridad con un producto autorizado en cuanto a la vía de administración, componente inactivo y en el mismo intervalo de concentración; b) Justificación del cambio del componente inactivo; c) Comparación de las propiedades físicas y químicas (pH, osmolaridad, toxicidad, etc); d) Información que demuestre que la modificación de los componentes inactivos no modifican esas propiedades.
Art. 9º — Cuando una Preparación Radiofarmacéutica de uso "in vivo" pueda ser utilizada con fines diagnósticos y terapéuticos, la misma deberá ser registrada de acuerdo a la reglamentación vigente para cada uno de los fines propuestos.
Art. 10. — Esta Administración Nacional de Medicamentos, Alimentos y Tecnología Médica se expedirá en un plazo de 120 (ciento veinte) días hábiles con respecto a la solicitud de autorización para la venta de productos para diagnóstico de uso "in vivo". Transcurrido el plazo, el interesado podrá solicitar a la Administración que se expida al respecto, aceptando o rechazando la petición.
Art. 11. — El referido plazo quedará automáticamente suspendido mientras se encuentre pendiente la entrega de documentación solicitada para la evaluación del producto.
Art. 12. — A los fines de verificar el cumplimiento de la presente Disposición, el Instituto Nacional de Medicamentos efectuará inspecciones con o sin retiro de muestras en los laboratorios elaboradores, fraccionadores y/o importadores, en los depósitos de los mismos, depósitos aduaneros, distribuidores y bocas de expendio.
Art. 13. — La presente Disposición entrará en vigencia dentro de los 60 (sesenta) días corridos, contados a partir del día siguiente al de su publicación en el Boletín Oficial.
Art. 14. — Regístrese, anótese, comuníquese a quienes corresponda, publíquese en el Boletín Informativo, dése a la Dirección Nacional de Registro Oficial para su publicación, cumplido, archívese PERMANENTE. — Manuel R. Limeres.
ANEXO I