Administración Nacional de Medicamentos, Alimentos y Tecnología Médica
SALUD PUBLICA
Disposición 5358/2012
Apruébanse las Buenas Prácticas de Farmacovigilancia. Objetivos. Formularios.
Bs. As., 10/9/2012
VISTO la Ley N° 16.643, sus Decretos Reglamentarios Nros. 9.763/64 y
150/92 (t.o. Decreto 177/93), los Decretos Nros. 1.490/92 y 341/92, la
Resolución del ex MS y AS N° 706/93, las Disposiciones ANMAT Nros.
822/94, 2552/95, 3870/99 y 2438/00, la Circular ANMAT N° 008/09 y el
Expediente N° 1-47-13183-08-8 del Registro de esta Administración
Nacional de Medicamentos, Alimentos y Tecnología Médica; y
CONSIDERANDO:
Que como consecuencia de la epidemia de una malformación llamada
focomelia en recién nacidos causada por la talidomida en Europa, a
partir del año 1960, varios países comenzaron a “vigilar” los efectos
“no deseables” de los medicamentos y que en 1968 la Organización
Mundial de la Salud (OMS), en el marco del Programa Internacional para
la Monitorización de Medicamentos, propuso la formación de un centro de
Farmacovigilancia Internacional.
Que el Sistema Nacional de Farmacovigilancia fue creado por Resolución
del ex Ministerio de Salud y Acción Social N° 706/93 estableciendo que
la Farmacovigilancia es una herramienta indispensable para el control y
fiscalización de las especialidades medicinales ya que permite la
detección temprana de los efectos adversos graves y/o inesperados de
los medicamentos en la etapa de uso extendido de éstos.
Que esta Administración Nacional ha organizado una Red Nacional de
Farmacovigilancia que incorpora a Efectores Periféricos de reconocida
idoneidad en la temática y que el Sistema Nacional está organizado con
un Efector Central con sede en el Departamento de Farmacovigilancia de
la ANMAT y efectores periféricos en distintos puntos del país.
Que de acuerdo con lo establecido por el artículo 4° de la Resolución
ex MS y AS N° 706/93 el Sistema cuenta con un Comité de Honor y una
Comisión Nacional de Farmacovigilancia.
Que por Disposición ANMAT N° 822/94 se constituyó la Comisión Nacional
de Farmacovigilancia con carácter ad honorem con profesionales del
entonces Consejo Asesor Permanente y representantes de tres Cámaras de
Medicamentos.
Que en 1994 Argentina fue admitida como País-Miembro del Programa
Internacional de Monitoreo de Eventos Adversos de Medicamentos de la
Organización Mundial de la Salud (UMC = Uppsala Monitoring Centre).
Que por Disposición ANMAT N° 2552/95 se estableció la Farmacovigilancia
Intensiva como monitoreo sistemático de la aparición de eventos
adversos de un ingrediente farmacéutico activo durante toda la etapa de
prescripción.
Que por Disposición ANMAT N° 3870/99 se solicitó a los Laboratorios
Farmacéuticos que nombraran un profesional de enlace con ANMAT y por
Disposición ANMAT N° 2438/00 se establecieron las “Bases para la
Ampliación de la Participación de la Industria Farmacéutica en el
Sistema Nacional de Farmacovigilancia”, considerándose de primordial
importancia la incorporación de la Industria Farmacéutica a los fines
de la difusión de los conceptos de Farmacovigilancia entre los
profesionales de la Salud.
Que la definición de la Organización Mundial de la Salud de 2002 sobre
Farmacovigilancia aclara que es la ciencia y las actividades
relacionadas con la detección, evaluación, comprensión y prevención de
los efectos adversos de los medicamentos o cualquier otro problema
relacionado con ellos.
Que la Farmacovigilancia permite la implementación de alertas
sanitarias y medidas administrativas de regulación y control,
contribuyendo al desarrollo de prescripciones y dispensaciones más
racionales a través de recomendaciones sobre efectos adversos
notificados producidos por principios activos y/o excipientes.
Que la notificación de reacciones adversas por parte de los
laboratorios ayudaría a acrecentar el conocimiento sobre el
beneficio/riesgo de los medicamentos comercializados en Argentina.
Que la publicación de las Buenas Prácticas de Farmacovigilancia es
considerada como la actualización de la disposición ANMAT 2438/00 sobre
la participación de la Industria Farmacéutica en el Sistema Nacional de
Farmacovigilancia, acorde a los avances y nuevos conceptos de seguridad
de medicamentos y seguridad de los pacientes que los requieren,
reconocidos internacionalmente.
Que la participación de la Industria ha sido insuficiente y a partir
del año 2005 se comenzó con un programa de mayor acercamiento entre las
empresas farmacéuticas y farmacovigilancia con reuniones anuales y
recordatorios de las disposiciones vigentes.
Que es sabido que un buen servicio de gestión de la seguridad de
medicamentos y de la Farmacovigilancia es un requisito imprescindible
para la detección precoz de los riesgos asociados a medicamentos y la
prevención de reacciones adversas a medicamentos.
Que existe la necesidad de promover e identificar precozmente los
problemas relacionados con los medicamentos distribuidos y
comercializados, con el objetivo de prevenir y minimizar los daños a la
salud de los pacientes.
Que las Buenas Prácticas de Farmacovigilancia son un conjunto de
reglas, procedimientos operativos y prácticas establecidas que se deben
cumplir para asegurar la calidad e integridad de los datos producidos
por las notificaciones y en determinados tipos de investigaciones o
estudios sobre medicamentos.
Que así como otras “Buenas Prácticas” (de Manufactura, de Ensayos
Clínicos, de Laboratorio, etc.) en diferentes países como EEUU, Canadá,
Francia, Reino Unido, España, Brasil, se han dictado las Buenas
Prácticas de Farmacovigilancia.
Que la Organización Panamericana de la Salud, a través del Grupo de
Trabajo de Farmacovigilancia de la Red Panamericana de Armonización de
la Regulación Farmacéutica ha publicado las Buenas Prácticas de
Farmacovigilancia de las Américas (documento técnico N° 5 de la Red
PARF).
Que los principios contenidos en el referido documento han sido
receptados por esta Administración Nacional en la Circular N° 008/09,
oportunamente publicada en la página web de este organismo.
Que el aludido documento incluye disposiciones referidas al
funcionamiento de los centros de Farmacovigilancia Nacional y
Provinciales así como también criterios generales sobre
Farmacovigilancia aplicables a la industria farmacéutica.
Que en ese contexto resulta necesario disponer de información adicional
en sus diferentes formas acerca del proceso de Farmacovigilancia que
debe ser llevado a cabo por los laboratorios titulares del registro de
medicamentos y por los responsables de las notificaciones.
Que debido a su condición de medicamentos, al impacto en la salud
pública que posee la utilización de las vacunas en el control de las
enfermedades inmunoprevenibles y a la importancia de la vigilancia de
los eventos adversos supuestamente atribuibles a la vacunación e
inmunización es conveniente incluir en las Buenas Prácticas de
Farmacovigilancia, las Buenas Prácticas de Farmacovigilancia en Vacunas.
Que con el objeto de optimizar la presentación del Plan de Gestión de
Riesgo, el proceso de notificación de eventos adversos a medicamentos,
desvíos de calidad, faltas de efectividad, errores de medicación,
eventos adversos por uso de medicamentos fitoterápicos, productos
vegetales y/o preparados de drogas vegetales, eventos supuestamente
atribuibles a la vacunación e inmunización y la notificación por parte
de los pacientes es conveniente disponer de formularios especialmente
diseñados a tales efectos.
Que el Departamento de Farmacovigilancia y la Dirección de Asuntos Jurídicos han tomado la intervención de su competencia.
Que se actúa en ejercicio de las facultades conferidas por los Decretos Nros. 1490/92 y 425/2010.
Por ello,
EL INTERVENTOR DE LA ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA
DISPONE:
Artículo 1° — Apruébanse las
Buenas Prácticas de Farmacovigilancia que obran en el ANEXO I que forma
parte integrante de la presente disposición, las cuales serán de
cumplimiento obligatorio para los Titulares de Autorización de Registro
y Comercialización (TARC) de especialidades medicinales.
Art. 2° — Apruébanse los Formularios que, como Anexo II, forman parte integrante de la presente disposición.
Art. 3° — Apruébase el Glosario que, como Anexo III, forma parte integrante de la presente disposición.
Art. 4° — El incumplimiento de
lo establecido en la presente disposición hará pasible a los
infractores de las sanciones previstas en la Ley de Medicamentos N°
16.463 y el Decreto N° 341/92.
Art. 5° — La presente
disposición entrará en vigencia a los treinta (30) días corridos,
contados a partir del primer día hábil siguiente al de su publicación
en el Boletín Oficial.
Art. 6° — Regístrese, Dése a la
Dirección Nacional del Registro Oficial para su publicación.
Comuníquese a CAEMe, CILFA, COOPERALA, CAPEMVeL, CAPGEN, COFA y a otras
entidades representativas del sector y a las entidades y organizaciones
profesionales correspondientes. Cumplido, archívese PERMANENTE. —
Carlos A. Chiale.
ANEXO I
BUENAS PRACTICAS DE FARMACOVIGILANCIA (BPFVG)
INTRODUCCION
1. RESPONSABILIDADES E INSPECCIONES
1.1. Responsabilidades y papel de los Titulares de Autorización de Registro y Comercialización (TARC)
1.2. Responsabilidades del Responsable de Farmacovigilancia (RFV)
1.3. Organización del área de Farmacovigilancia (FVG) y miembros involucrados
1.4. Inspecciones de Farmacovigilancia
2. INFORMES PERIODICOS DE ACTUALIZACION SEGURIDAD (IPAS)
2.1. Introducción
2.2. Periodicidad
2.3. Selección e inclusión de datos
2.4. Poblaciones especiales
2.5. Control de calidad
2.6. Modelo de IPAS
3. PLAN DE GESTION DE RIESGO (PGR)
3.1. Introducción
3.2. Descripción y requerimientos
3.3. Especificaciones de seguridad
3.4. Plan de Farmacovigilancia
3.5. Evaluación de las necesidades de actividades de minimización de riesgo
3.6. Plan de Minimización de Riesgos
3.7. Actividades de Minimización de Riesgos
3.8. Asegurar la efectividad de las actividades de minimización de riesgo
3.9. Resumen de las actividades
3.10. Presentación de documentos actualizados
4. BUENAS PRACTICAS DE FARMACOVIGILANCIA EN VACUNAS
4.1. Introducción
4.2. Eventos supuestamente atribuibles a la vacunación e inmunización (ESAVI)
4.3. Responsabilidades ANMAT - Farmacovigilancia
4.4. Investigación de los eventos supuestamente atribuibles a la vacunación e inmunización (ESAVI)
4.5. Responsabilidades del Titular de Autorización de Registro y Comercialización (TARC).
INTRODUCCION
La Salud Pública, a través de la Farmacovigilancia, logra identificar,
cuantificar, evaluar y prevenir los posibles riesgos derivados del uso
de los medicamentos durante la etapa de post-comercialización.
Específicamente, la Farmacovigilancia es una actividad de
responsabilidad compartida entre los agentes que utilizan el
medicamento, es decir, los Titulares de la Autorización de Registro y
Comercialización (industria farmacéutica), la autoridad sanitaria
regulatoria (ANMAT, Departamento de Farmacovigilancia), los
profesionales de la salud (médicos, farmacéuticos, etc.) y los
pacientes. El Titular de una Autorización de Registro y
Comercialización (TARC) debe velar por la existencia de un Sistema de
Farmacovigilancia que le permita asumir sus responsabilidades y
obligaciones con relación a las especialidades medicinales que
comercializa y asegurar el cumplimiento de las medidas y procedimientos
oportunos, en tiempo y forma. A fin de facilitar el desarrollo y
actualización de estas obligaciones, se establecen las Buenas Prácticas
de Farmacovigilancia para la industria farmacéutica, consistentes en
estándares de calidad referentes a la organización y funcionamiento de
los Titulares de Autorización de Registro y Comercialización de
medicamentos. El cumplimento de estos principios tiene el objetivo de
garantizar la autenticidad y la calidad de los datos de seguridad, para
la evaluación continua de los riesgos asociados a las especialidades
medicinales que comercializa. Es deseable que los beneficios y
conocimientos aportados en los capítulos siguientes sirvan, además, de
herramienta para todos los profesionales involucrados en
Farmacovigilancia.
1. RESPONSABILIDADES E INSPECCIONES
1.1. RESPONSABILIDADES DEL TITULAR DE LA AUTORIZACION DE REGISTRO Y COMERCIALIZACION (TARC)
La responsabilidad legal derivada del cumplimiento de las obligaciones
de Farmacovigilancia recae siempre en el TARC, debiendo asumir sus
responsabilidades y obligaciones con respecto a las especialidades
medicinales que tiene autorizadas y asegurar la adopción de las medidas
oportunas cuando sea necesario. En conformidad con estas Buenas
Prácticas de Farmacovigilancia cada laboratorio Titular de la
Autorización de Registro y Comercialización deberá:
a) Llevar un registro detallado de todas las sospechas de reacciones
adversas que se produzcan con sus especialidades medicinales en
Argentina. Las notificaciones de sospechas de casos individuales de
reacciones adversas que cumplan los criterios señalados se comunicarán
preferentemente por vía electrónica utilizando la terminología médica
internacionalmente aceptada.
b) Registrar y comunicar las sospechas de reacciones adversas serias
ocurridas en Argentina al Departamento de Farmacovigilancia de la
ANMAT. Dicha notificación debe realizarse dentro de los 15 días
corridos siguientes a la recepción de la información. En caso de muerte
o amenaza de vida la notificación deberá realizarse dentro de los 7
días corridos siguientes a la recepción de la información.
c) Notificar al Departamento de Farmacovigilancia de la ANMAT cuando se
detecte que una mujer embarazada es expuesta a un medicamento, a pesar
de no producirse un evento adverso.
d) Notificar tanto las sospechas de reacciones adversas serias
asociadas a medicamentos autorizados, como las de aquellos que se hayan
utilizado en condiciones diferentes a las autorizadas. Las
notificaciones de reportes no serios deberán enviarse por vía
electrónica de manera bimestral en el formato establecido por esta
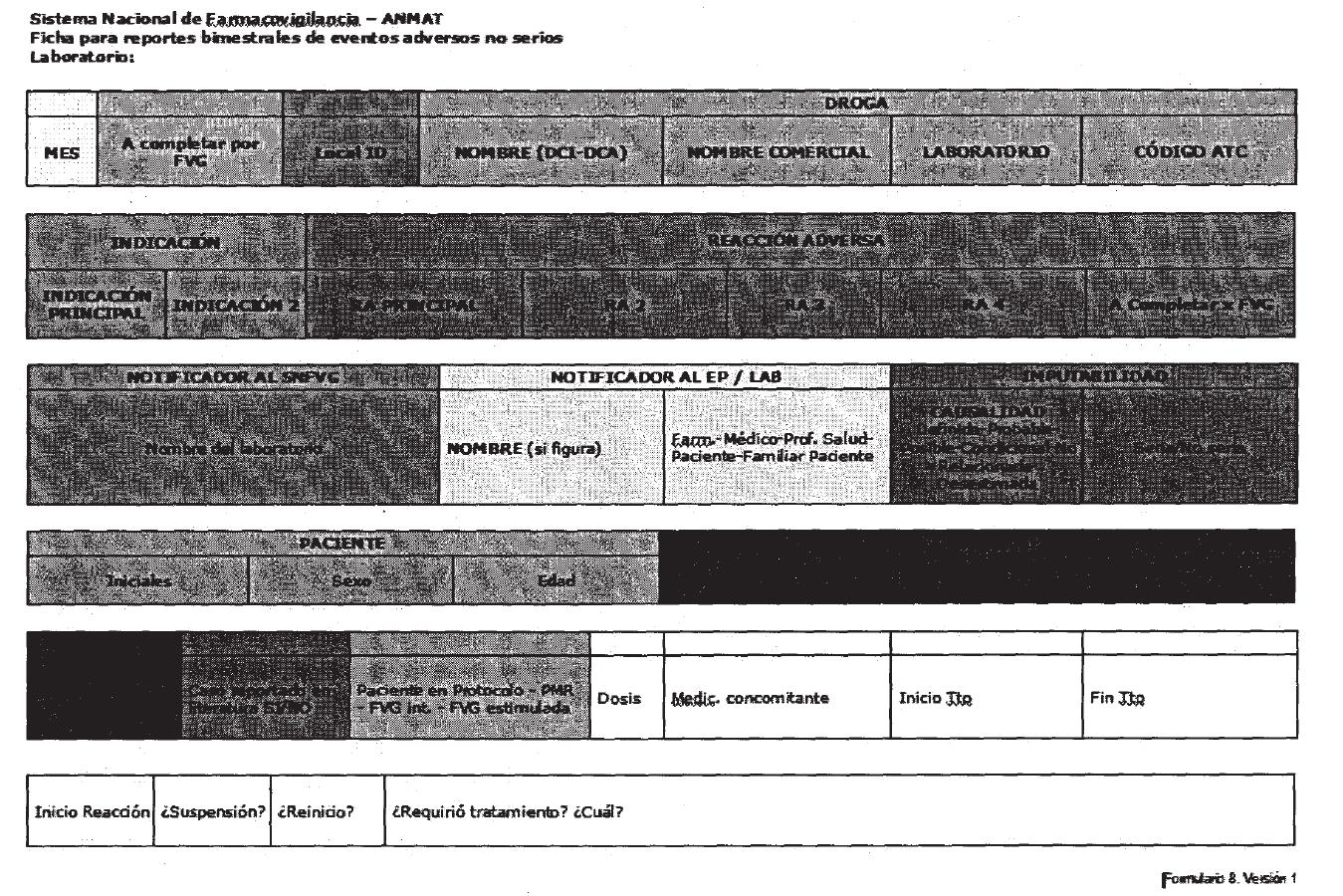
Administración (ver Formulario 8 del Anexo II).
e) Asegurarse de que los reportes enviados tengan la mínima información
indispensable. Dicha información consiste en: la fecha de comienzo del
evento adverso, la fecha de comienzo de la administración de la
medicación sospechosa, la edad del paciente, la descripción del evento
completa y el nombre de la droga o drogas implicadas (DCA —Denominación
Común Argentina— y marca).
f) Realizar un seguimiento de la bibliografía científica nacional e
internacional, con el fin de identificar los casos publicados de
reacciones adversas ocurridas en Argentina con sus especialidades
medicinales.
g) Garantizar que las sospechas de reacciones adversas serias que
ocurran durante el transcurso de un estudio post-autorización, y de las
que pueda esperarse que razonablemente tengan conocimiento, sean
comunicadas utilizando los criterios especificados en el párrafo b)
siempre y cuando la droga se encuentre registrada en nuestro país.
h) Presentar ante el Departamento de Farmacovigilancia de la ANMAT
todas las sospechas de reacciones adversas nacionales e internacionales
en un Informe Periódico de Actualización de Seguridad (IPAS). Este
informe, cuando se trate de productos de laboratorios multinacionales o
de productos licenciados a laboratorios nacionales, es denominado PSUR.
según sus siglas en inglés (Periodic Safety Update Report) y suele ser
redactado en forma globalizada, concluyendo si se requiere alguna
modificación en el prospecto o en alguna característica de
comercialización. En estos casos, deberá presentarse exclusivamente el
“resumen”. Dicha presentación deberá efectuarse inmediatamente frente a
un requerimiento de la ANMAT y, asimismo, de forma periódica con
arreglo a los plazos previstos internacionalmente, siempre que no se
hayan establecido otros requisitos como condición para conceder la
autorización de registro del medicamento. En caso de especialidades
medicinales que sólo son comercializadas en nuestro país, los
laboratorios deberán confeccionar un Informe Periódico de Actualización
de Seguridad (IPAS) con los lineamientos establecidos por esta
Administración y presentarlos para todas sus especialidades medicinales
comercializadas en cualquiera de las modalidades. En caso de productos
innovadores (moléculas nuevas, nuevas indicaciones, etc.) el TARC podrá
solicitar la modificación de la periodicidad.
i) En todos los casos del ítem anterior deberán incluirse datos
registrales y de seguridad locales, para el período y el producto que
se está informando, tales como:
• La historia de la especialidad medicinal en Argentina desde su
registro (fecha de registro, fecha de inicio de comercialización,
modificaciones post-aprobación, etc. y los respectivos números de
disposición autorizantes).
• Número de unidades vendidas en Argentina (en cualquier modalidad de
venta que se trate, aclarando si es por año o por mes) y estimación de
la cantidad de pacientes expuestos en nuestro país (y la forma en que
se realizó el cálculo de dicha estimación).
• Notificaciones de eventos adversos, serios y no serios, desvíos y
problemas de calidad/falta de eficacia ocurridos en el país, así como
también reclamos de pacientes, retiros de mercado, alertas, comunicados
o cualquier otra situación vinculada a la seguridad del producto
ocurrida en Argentina y en otros países para la especialidad medicinal
en cuestión.
• La existencia de un Plan de Gestión de Riesgos, las modificaciones
eventuales al Plan o la necesidad de implementarlo en caso de que aún
no se lleve a cabo.
• Modificaciones en la información de seguridad o en los prospectos,
presentada o pendiente de presentación ante ANMAT. Si están pendientes
de presentación: informar fecha prevista. Si están presentadas pero aún
no aprobadas: informar número de expediente. Si están presentadas y
aprobadas: informar número de disposición autorizante.
j) Realizar una evaluación continua de la relación beneficio-riesgo de
los medicamentos que tenga autorizados en Argentina y comunicar
inmediatamente a la ANMAT toda aquella nueva información que pueda
influir en la evaluación global de la relación beneficio-riesgo.
k) Suministrar un informe de la relación beneficio-riesgo cuando la ANMAT lo solicite.
l) Llevar a cabo los Planes de Gestión de Riesgo que para cada
medicamento se establezcan, incluyendo, los estudios que las
autoridades competentes juzguen necesarios para evaluar la seguridad
del medicamento.
m) Abstenerse de comunicar al público y a los profesionales de la salud
cuestiones de Farmacovigilancia relativas a su medicamento autorizado
sin que previamente se hayan comunicado tales cuestiones, con al menos
24 horas de antelación, a la ANMAT. El TARC se cerciorará de que la
información se presente de manera objetiva y no sea engañosa, y sin
omitir información de seguridad relevante.
n) Reportar las sospechas de eventos adversos en vacunas (ESAVI), tanto
de vacunas incluidas en el Calendario Nacional de Vacunación como
aquellas fuera de calendario, acorde al párrafo b), con la excepción de
aquellas incluidas en Farmacovigilancia activa. Los eventos serios e
inesperados de vacunas de calendario deberán ser, además, enviados al
Programa Nacional de Control de Enfermedades Inmunoprevenibles,
dependiente de la Dirección de Epidemiología del Ministerio de Salud de
la Nación.
o) Cumplimentar las disposiciones de los programas de Farmacovigilancia Intensiva vigentes.
p) Disponer en Argentina de una persona adecuadamente calificada como
Responsable en materia de Farmacovigilancia (RFV) y comunicarlo al
Departamento de Farmacovigilancia de la ANMAT.
q) Garantizar que todo el personal de la compañía tenga formación
adecuada a sus responsabilidades en materia de Farmacovigilancia.
r) Garantizar el cumplimiento de los procedimientos de trabajo
apropiados, basándose en los documentos (POEs - Procedimientos
Operativos Estándar) aprobados.
s) Garantizar un sistema de archivo que permita conservar adecuadamente
toda la documentación relacionada con las responsabilidades y
actividades de Farmacovigilancia. Las responsabilidades en la gestión
del archivo tienen que estar definidas por escrito.
t) Establecer un programa de auditorías, con el fin de garantizar que
el sistema de Farmacovigilancia se adecue a las Buenas Prácticas de
Farmacovigilancia.
1.2. RESPONSABILIDADES DEL RESPONSABLE DE FARMACOVIGILANCIA (RFV)
El RFV será el único interlocutor válido en términos de
Farmacovigilancia ante la ANMAT - Departamento de Farmacovigilancia.
Por lo tanto, el RFV debe presentarse a las autoridades sanitarias
competentes, como miembro responsable del área, proporcionando su
nombre, teléfonos y dirección electrónica de contacto. El TARC
facilitará al RFV toda la información referente a la seguridad de cada
especialidad farmacéutica autorizada, convenientemente actualizada. El
RFV garantizará el cumplimiento de las siguientes actividades:
a) Crear y mantener un sistema para recopilar, tratar y evaluar la
información sobre todas las sospechas de reacciones adversas
notificadas al personal de la empresa y a los agentes de propaganda
médica (APM).
b) Notificar sospechas de reacciones adversas (SRA), cumpliendo los plazos previamente descriptos.
c) Elaborar y/o revisar los Informes Periódicos de Actualización de Seguridad (IPAS).
d) Responder, en tiempo y forma, cualquier petición de información de
las autoridades competentes en materia de seguridad de medicamentos.
e) Evaluar en forma permanente la relación beneficio-riesgo durante el
período de post-autorización, y comunicar inmediatamente a las
autoridades competentes cualquier información que pudiera suponer un
cambio en dicha relación.
f) Asegurar los mecanismos necesarios para cumplir con las medidas
regulatorias establecidas para los medicamentos de cuya
Farmacovigilancia es responsable, así como todas aquellas medidas y
estudios incluidos en el Plan de Gestión de Riesgo y que se prevean
realizar en nuestro país.
g) Establecer criterios de identificación y de valoración de la gravedad de las señales de alerta.
h) Revisar periódicamente la literatura científica sobre sospechas de
reacciones adversas (SRA) de los ingredientes farmacéuticos activos de
los que el TARC es titular.
i) Actuar como punto de contacto para las inspecciones de Farmacovigilancia realizadas en Argentina.
1.3. ORGANIZACION DEL AREA DE FARMACOVIGILANCIA/MIEMBROS INVOLUCRADOS
a) El TARC debe poner a disposición del RFV los recursos humanos y
materiales necesarios para llevar a cabo de manera adecuada sus
responsabilidades.
b) El TARC debe disponer de un organigrama actualizado que refleje la
relación jerárquica que hay entre el RFV, la dirección y el resto de
departamentos del TARC.
c) El RFV debe tener una formación adecuada y experiencia en
Farmacovigilancia. Si no tiene calificación médica, debe tener acceso a
una persona que tenga esa calificación dentro del equipo de trabajo, ya
que la terminología médica deberá ser interpretada y analizada
correctamente.
d) Debe haber una persona designada como suplente en caso de ausencia
del RFV. Esta persona debe tener la formación adecuada en el Area.
e) El personal de Farmacovigilancia debe conocer las funciones y
responsabilidades asignadas, las cuales tienen que estar especificadas
por escrito, en las descripciones de los puestos de trabajo, aprobados
por la Dirección.
f) El TARC debe mantener un registro actualizado con los títulos de
cada integrante, el currículum vitae, la descripción del puesto de
trabajo y la capacitación del personal involucrado en las tareas de
Farmacovigilancia.
g) El Director Técnico del laboratorio, como responsable ante la ANMAT,
firmará la documentación presentada ante el Departamento de
Farmacovigilancia.
1.4. INSPECCIONES EN FARMACOVIGILANCIA
Para asegurar que el TARC cumplimente las obligaciones regulatorias en
Farmacovigilancia, la ANMAT conducirá inspecciones de
Farmacovigilancia. Los resultados de las inspecciones serán provistos
al TARC, que tendrá la oportunidad de formular las consideraciones que
hagan a su derecho. Estos resultados serán utilizados para mejorar el
cumplimiento de las BPFVG por parte del TARC y podrían ser usados
también como la base de acciones regulatorias.
1.4.1. Inspecciones de rutina
a) El objetivo de estas inspecciones es determinar si el TARC posee
personal, sistemas y recursos en funcionamiento en consonancia con las
obligaciones regulatorias. Estas inspecciones pueden ser requeridas
para uno o más medicamentos específicos elegidos como ejemplos para
verificar el funcionamiento del sistema de Farmacovigilancia del TARC y
el cumplimiento de sus obligaciones.
b) Las inspecciones de rutina serán efectuadas cuando:
• El TARC no fue previamente inspeccionado.
• El TARC ha comercializado su primer medicamento en Argentina.
• El TARC está involucrado en un proceso de fusión.
• El TARC ha cambiado su sistema significativamente (por ej., nueva base de datos).
• La ANMAT lo considere pertinente.
1.4.2. Inspecciones dirigidas
Las inspecciones dirigidas serán efectuadas cuando se reconozca una
situación específica de seguridad o falta de cumplimiento y la ANMAT
determine que una inspección es la mejor manera de proceder. Estas
situaciones pueden ser:
• Demoras o fallas en el cumplimiento de obligaciones específicas o
medidas de seguimiento relacionadas al monitoreo de seguridad de
medicamentos.
• Demoras en notificaciones expeditivas o periódicas.
• Presentación incompleta o de mala calidad de los IPAS.
• Inconsistencias entre notificaciones y otras fuentes de información.
• Cambios en el balance beneficio-riesgo de algún medicamento.
• Experiencias previas de inspección.
• Información incompleta de los seguimientos requeridos por ANMAT.
• Comunicación de información de Farmacovigilancia al público en general sin notificar previamente a la ANMAT.
• Retiro de medicamentos por motivos de seguridad sin previo aviso a la ANMAT.
• Cuando la ANMAT lo considere pertinente.
1.4.3. Inspecciones no anunciadas
Está previsto que la mayoría de las inspecciones sean anunciadas. Sin
embargo, en ocasiones, será apropiado conducir inspecciones no
anunciadas o anunciadas con poco anticipo.
1.4.4. Temas a revisar durante la inspección de Farmacovigilancia
Se revisarán, aunque no exclusivamente, los siguientes ítems como parte de un plan de inspección.
a) Aspectos legales y administrativos
• La documentación de los responsables de las actividades de Farmacovigilancia.
• Identificación del RFV en el TARC.
• Disponibilidad inmediata de la información sobre las sospechas de
eventos adversos y otras notificaciones de FVG, así como también
Informes Periódicos de Actualización de Seguridad, Planes de Gestión de
Riesgo u otra información que corresponda.
• Cumplimiento de la obligación de informar a la ANMAT los eventos adversos en relación con los productos autorizados.
• Requerimientos especiales para reportar eventos adversos a la ANMAT o
para el monitoreo de seguridad post-autorización, por ej., Planes de
Gestión de Riesgo.
• Preparación y presentación de Informes Periódicos de Actualización de Seguridad.
• Recolección y reporte de notificaciones espontáneas de eventos adversos.
• Recolección, seguimiento y notificación de exposición durante el embarazo.
• Recolección, seguimiento y notificación de exposición de la población pediátrica.
• Puesta a disposición de las autoridades competentes de cualquier otra
información pertinente a la evaluación de los riesgos y beneficios de
un medicamento.
b) Estructura organizativa y gestión de la información
b.1) Sistema de calidad y procedimientos operativos estándar (POEs) para las actividades de Farmacovigilancia.
La documentación de los POEs y las instrucciones para cubrir todos los
aspectos de la Farmacovigilancia deben incluir, pero no se limitan a,
las siguientes actividades:
• Recolección y gestión de datos de Farmacovigilancia (de los
pacientes, profesionales de la salud, los departamentos de información
médica, los departamentos de reclamos de calidad, los departamentos de
asuntos regulatorios, departamentos jurídicos, centros de producción,
etc.).
• Evaluación de la causalidad.
• Determinación de la seriedad de las reacciones adversas y si están listadas.
• Codificación.
• Evitar la duplicación de informes.
• Asegurar el cumplimiento de la presentación de informes.
• Identificación y seguimiento de los informes iniciales.
• Utilización de reportes de otras organizaciones (por ej., de Organizaciones de Investigación por Contrato).
• Asegurar la integridad de la información contenida en las bases de datos.
• Revisión, validación y seguimiento de sospechas de reacciones adversas.
• Gestión de datos: almacenamiento apropiado y recuperación de
información, seguimiento de informes y cumplimiento de la
confidencialidad.
• Reportes expeditos a la ANMAT.
• Seguimiento de la literatura científica en todo el mundo.
• Elaboración y presentación de Informes Periódicos de Actualización de Seguridad (IPAS).
• Gestión de solicitudes de información por las autoridades competentes.
• Gestión de las restricciones de seguridad urgentes.
• Actualización de información de seguridad y de prospectos.
• Detección de señales.
• Comunicación con las autoridades competentes según sea necesario.
• Elaboración y presentación de los Planes de Gestión de Riesgo (PGR).
• Listas de organización para identificar al personal clave.
• Control de los POEs y otros documentos de procedimientos, incluyendo
la escritura, revisión, aprobación, actualización, distribución y
ejecución.
• Revisión de los documentos y procesos de acciones correctivas y preventivas.
• Manejo de información de seguridad proveniente de otros departamentos
de la compañía, que puede incluir, pero no limitarse a, información
médica, legal, regulatoria, de comercialización, de producción, desvíos
de calidad, reclamos.
• Auditoría del sistema de Farmacovigilancia:
- Realización de auditorías de Farmacovigilancia.
- Manejo de los hallazgos de auditoría y los procesos de comunicación.
- Capacitación y entrenamiento de los auditores.
b.2) Responsable de Farmacovigilancia (RFV)
• Documentación que identifique al RFV junto con su calificación y entrenamiento.
• Documentación del RFV y datos de contacto.
• Verificación de que el RFV tiene acceso a toda la información de Farmacovigilancia y de seguridad de medicamentos.
• Verificación de que el RFV ha sido notificado a la ANMAT.
• Verificación de que el RFV tiene suficiente autoridad para hacer
modificaciones al sistema de Farmacovigilancia con el fin de asegurar
el cumplimiento.
• Documentación para la delegación de tareas.
• Verificación del proceso de respaldo (back-up) cuando el RFV está ausente.
b.3) Recursos y capacitación del personal
• Entrevista del personal involucrado en cualquier actividad de
Farmacovigilancia, incluyendo, entre otras, a las áreas médica, de
información al público, de asuntos regulatorios, de asuntos legales y
departamento de calidad.
• Documentación con la descripción del trabajo, calificación y
capacitación de quienes participan en cualquier actividad de
Farmacovigilancia.
• Documentación sobre las políticas y procedimientos para la formación del personal.
1.4.5. Clasificación de incumplimientos
Los incumplimientos y deficiencias encontrados durante las inspecciones
de Farmacovigilancia se clasifican de la siguiente forma:
a) Críticos. Deficiencias en el Sistema de Farmacovigilancia, prácticas
o procesos, que afecten en forma adversa los derechos, la seguridad o
el bienestar de los pacientes o que presenten un riesgo para la salud
pública o que representen una violación seria a la legislación vigente.
b) Mayores. Deficiencias en el Sistema de Farmacovigilancia, prácticas
o procesos que potencialmente podrían afectar en forma adversa los
derechos, la seguridad o el bienestar de los pacientes o que pudieran
implicar un riesgo para la salud pública o que representen una
violación a la legislación vigente.
c) Otros. Deficiencias en el Sistema de Farmacovigilancia, prácticas o
procesos para los que no se espera que afecten en forma adversa los
derechos, la seguridad o el bienestar de los pacientes.
1.4.6. Medidas regulatorias
Las siguientes acciones pueden resultar de una inspección de Farmacovigilancia:
• Orientación: recomendaciones sobre el cumplimiento de los requerimientos regulatorios establecidos en la presente disposición.
• Reinspección: nuevas inspecciones para verificar el cumplimiento de
las orientaciones o de los requerimientos regulatorios establecidos en
la presente disposición.
• Advertencia: recomendaciones formales al TARC sobre la necesidad de cumplir con los requerimientos regulatorios.
• Restricciones por motivos de seguridad (tales como suspensión de
comercialización y uso o cancelación del registro de un producto).
• Otras medidas que la ANMAT considere pertinentes de acuerdo con la normativa aplicable.
1.4.7. Seguimiento de los resultados de la inspección
Cuando la inspección resulte en faltas de cumplimiento, se le
solicitará al TARC un plan para corregir los incumplimientos y evitar
la recurrencia. Se le podrá requerir al TARC reportes y, cuando sea
necesario, la evidencia del progreso. Podrá haber reinspecciones en un
tiempo apropiado para verificar el progreso y el éxito de estas
acciones.
2. INFORMES PERIODICOS DE ACTUALIZACION DE SEGURIDAD (IPAS)
2.1. Introducción
Los Informes Periódicos de Actualización de Seguridad son documentos
donde se presentan todos los datos de Farmacovigilancia de un
medicamento obtenidos en un determinado período, establecido a partir
de su fecha de comercialización. El objetivo de estos informes es que
los laboratorios farmacéuticos participen en la recolección de datos y
de notificaciones, evalúen la información de seguridad reunida y la
presenten de manera estandarizada a la Autoridad Regulatoria que ha
registrado el medicamento. Tienen que brindar la experiencia nacional e
internacional sobre la seguridad de un medicamento con los objetivos de:
• Comunicar toda la nueva información relevante sobre seguridad procedente de fuentes adecuadas.
• Presentar de forma resumida la situación de la Autorización de
Registro y Comercialización en distintos países, siempre que
corresponda, y cualquier modificación importante relacionada con la
seguridad.
• Facilitar periódicamente la oportunidad de reevaluación de la
relación beneficio/riesgo y de decidir si se modifica la información
terapéutica y de seguridad de la especialidad medicinal.
Se informarán las medidas adoptadas utilizando canales de comunicación apropiados, tales como:
• El rotulado oficial establecido (envase primario, envase secundario,
literatura interior o prospecto, hojas informativas, monografía,
resumen de las características del producto, etc.)
• Cartas de respuesta a quejas y reclamos.
• Comunicaciones de seguridad dirigidas a profesionales sanitarios.
• Medidas sanitarias de reducción de riesgos.
• Boletines impresos o distribuidos por correo electrónico o mediante la utilización de internet.
• Artículos científicos.
• Advertencias públicas en medios de difusión masiva (prensa escrita, radio, televisión o en Internet).
Es responsabilidad del RFV la presentación de manera adecuada y en
tiempo de los IPAS. Su preparación puede ser delegada a otra persona
adecuadamente entrenada.
2.2. Periodicidad
2.2.1. Periodicidad estándar
A partir de la fecha de comercialización, el TARC deberá presentar los IPAS de acuerdo a la siguiente periodicidad:
• Semestralmente durante los dos primeros años tras el inicio de la comercialización.
• Anualmente, en los dos años siguientes.
• Cada tres años a partir del 5º año, o según se establezca.
2.2.2. Periodicidad en casos especiales
En algunas situaciones particulares, el TARC deberá presentar los IPAS:
• Con la periodicidad establecida en los Planes de Gestión de Riesgo,
en los Programas de Farmacovigilancia Intensiva u en otras modalidades
de Farmacovigilancia que lo requieran, en caso de corresponder.
• Inmediatamente a partir de la solicitud de la autoridad regulatoria.
El TARC podrá presentar los IPAS de acuerdo a los plazos
internacionales, en el caso de productos comercializados por
laboratorios multinacionales o de productos licenciados a laboratorios
nacionales, previo aviso a la autoridad regulatoria local.
La mayoría de los productos similares pueden ser ubicados en el mismo
ciclo que el innovador. En la mayoría de los casos, esto será en el
período trianual. Sin embargo, la periodicidad estándar se aplicará
desde el inicio para especialidades medicinales de origen biológico con
antecedentes, para medicamentos con cuestiones de seguridad específicas
o de vigilancia intensiva (isotretinoína, clozapina, talidomida, etc.)
y para nuevas formulaciones, vías de administración o indicaciones.
El TARC podrá solicitar la modificación de las periodicidades arriba citadas, justificando debidamente dicha solicitud.
2.3. Selección e inclusión de datos
Las fuentes de información de reacciones adversas ocurridas con la especialidad medicinal incluyen:
• Reportes directos al TARC. Esto incluye reportes o notificaciones
espontáneas, notificaciones obtenidas a través de modalidades activas
de Farmacovigilancia, estudios realizados por el TARC, notificaciones
de pacientes al TARC, etc.
• Reportes de reacciones adversas provenientes de la literatura.
• Reportes recibidos por autoridades regulatorias a nivel nacional y mundial.
• Reportes de otras fuentes (tales como otras compañías, registros
varios, centros de toxicología o bases de datos epidemiológicos).
El TARC debe tener procedimientos formales para asegurarse de que todos
los casos pertinentes sean incluidos en el IPAS. Los casos relevantes
de ensayos clínicos, incluyendo reacciones adversas medicamentosas
serias e inesperadas (RAMSI) o reacciones adversas medicamentosas
serias (RAMS) sospechados durante el período reportado deben ser
incluidos en el IPAS. Toda la información de seguridad concerniente a
un producto que provenga de un estudio (clínico, no clínico y
epidemiológico) durante el período cubierto debe discutirse si de
alguna manera altera el perfil beneficio-riesgo.
2.4. Poblaciones especiales
La información de seguridad para la población pediátrica, mayores de 65
años y embarazadas deberá ser siempre incluida y analizada en los IPAS.
Si no hubiese, o hubiese muy poca información sobre estas poblaciones,
deberá reportarse en el IPAS de cualquier manera. La adición de una
indicación pediátrica a un medicamento ya comercializado modificará la
periodicidad, debiendo presentarse los IPAS semestralmente durante los
próximos dos años. Estos reportes deberán prestar atención al uso
pediátrico.
2.5. Control de Calidad
Dado que el RFV es responsable de la redacción del IPAS, debe
asegurarse de que existan procedimientos para controlar que los datos
presentados en estos reportes sean exactos y completos.
2.6. Modelo de IPAS
Sin perjuicio de que puedan ser tenidos en cuenta otros formatos
internacionalmente aceptados (por ej., informes periódicos de
evaluación riesgo-beneficio), en esta sección se describe un modelo de
IPAS, con guías para su realización y los contenidos a incluir.
2.6.1. Resumen
El TARC debe preparar un resumen ejecutivo de cada IPAS que contenga
una descripción de la información más importante. Esta sección debe
estar en el inicio del IPAS y debe incluir información resumida de lo
siguiente:
• El estatus de autorización mundial del producto (incluyendo un
listado de países donde el producto está autorizado/comercializado) y
las indicaciones autorizadas.
• Otra información relevante regulatoria relacionada al período
cubierto en el IPAS (por ej., cualquier información de restricción de
uso debe ser resaltada).
• Datos de exposición de pacientes.
• Número de nuevos casos recibidos durante el período cubierto por el IPAS. y los números de casos acumulados.
• Situaciones particulares de seguridad investigadas.
• Conclusiones finales del IPAS.
• Cuando el TARC haya realizado una revisión de una o más situaciones
de seguridad, esto debe estar descripto en el resumen del IPAS.
2.6.2. Introducción
El TARC debe presentar el IPAS como un documento integral y único, pero
a la vez teniendo en cuenta los IPAS previos. Se debe hacer referencia
al (los) producto(s) incluidos en el IPAS, y a los excluidos. Estas
exclusiones deben ser explicadas, por ejemplo, si son cubiertas en otro
IPAS (como en el caso de un producto combinado). Si es sabido que el
IPAS del mismo producto será presentado por otro TARC debe ser
considerada la posibilidad de presentar datos duplicados.
2.6.3. Estatus de autorización mundial
Esta sección del IPAS provee información acumulada respecto de medidas
regulatorias de comercialización en todos los países, en relación a lo
siguiente:
• Fechas de autorización y renovación subsiguientes.
• Cualquier modificación de la autorización, si es relevante para la seguridad.
• Indicaciones de tratamiento y poblaciones especiales cubiertas cuando sea relevante.
• Rechazo de la aprobación, incluyendo explicación, por las autoridades regulatorias específicas.
• Desistimiento de la solicitud de registro por parte de la compañía, si está relacionada con la seguridad o la eficacia.
• Fechas de inicio de comercialización.
• Fechas en que la autorización de comercialización ha sido suspendida
o revocada por la autoridad regulatoria o voluntariamente por el TARC.
• Nombres comerciales.
La información debe ser provista preferentemente en forma tabulada.
Típicamente, las indicaciones de uso, las poblaciones tratadas y las
dosis son iguales en la mayoría de los países donde el medicamento se
comercializa. Sin embargo, cuando haya diferencias importantes que
reflejen los diferentes tipos de exposición de pacientes, esta
información debe explicitarse.
2.6.4. Actualización de acciones tomadas por la autoridad regulatoria o por el TARC por razones de seguridad
Esta sección debe incluir detalles de los siguientes tipos de medidas
relacionadas con la seguridad del producto tomadas durante el período
cubierto por el IPAS, ocurridas nacional e internacionalmente.
• Retiro, suspensión o cancelación del certificado de registro del producto.
• Imposibilidad de obtener la reinscripción del certificado de registro del producto.
• Restricciones en la distribución, uso, condición de venta, etc.
• Suspensión de ensayos clínicos.
• Modificación de las dosis.
• Cambios en las poblaciones tratadas o indicaciones.
• Cambios en la formulación.
• Restricciones de seguridad.
2.6.5. Cambios en la información de referencia de seguridad
Cambios en la información de seguridad, tales como nuevas
contraindicaciones, precauciones, advertencias, reacciones adversas o
interacciones, realizados durante el período cubierto por el IPAS deben
describirse claramente, con presentación de las secciones modificadas.
2.6.6. Pacientes expuestos
Cuando sea posible, debe presentarse un cálculo preciso del número de
pacientes expuestos, el cual debe cubrir el mismo período que el IPAS.
Cuando resulte muy dificultoso obtener y validar datos precisos de
exposición, debe aportarse el estimado de los pacientes, junto con el
método utilizado para realizar el cálculo. Debe presentarse una
justificación si el número preciso de pacientes es imposible de
obtener. En su lugar, pueden utilizarse otras medidas de exposición,
tales como pacientes-días, número de prescripciones, número de unidades
vendidas, etc. De esta manera, dada la dificultad para realizar las
estimaciones, la información de los pacientes expuestos puede también
ser provista como paciente - tiempo de exposición (días-meses-años).
Asimismo, se puede utilizar el concepto de Dosis Diaria Definida (DDD)
para arribar a otros estimados. Cuando los datos de pacientes expuestos
estén basados en información de un período que no cubre completamente
el período del IPAS, el TARC podrá, justificando adecuadamente,
extrapolar usando los datos disponibles. Cuando sea relevante, la
información sobre exposición debe estratificarse de acuerdo con
variables significativas tales como edad, indicación, dosis y duración
de tratamiento.
2.6.7. Presentación de casos
2.6.7.1. Análisis de casos individuales
Esta sección debe incluir una descripción y análisis de casos
seleccionados conteniendo nueva o relevante información de seguridad.
Se proveerá una descripción del criterio utilizado para seleccionar los
casos.
2.6.7.2. Casos presentados como listados
Los casos deben ser incluidos en listados. Se debe intentar evitar
duplicados de la literatura y de fuentes regulatorias. Estos casos
incluyen:
• Todas las reacciones adversas serias y no serias (no listadas) de
reportes espontáneos, de la literatura o de las autoridades
regulatorias específicas.
• Todas las reacciones adversas serias que provengan de estudios
postcomercialización y otros (incluyendo los que son parte del Plan de
Gestión de Riesgo).
Además los siguientes casos se presentarán como anexo al IPAS:
• Reacciones serias y no serias listadas espontáneas.
Los listados deben incluir cada paciente solamente una vez, sin
importar cuántas reacciones adversas hayan sido reportadas para el
caso. Si hay más de una reacción, deben ser todas mencionadas, pero el
caso debe ser listado según la reacción adversa más seria a ser
determinada por el TARC. Es posible que el mismo paciente experimente
reacciones adversas diferentes en distintos momentos. Estas situaciones
serán tratadas como reportes separados. En estas circunstancias, el
mismo paciente será incluido en el listado más de una vez.
El listado deberá incluir los siguientes encabezados:
• Número de caso asignado por el TARC.
• País en que el caso ocurrió.
• Fuente (por ej., ensayo clínico, literatura, espontáneo, autoridad regulatoria).
• Edad y sexo de pacientes.
• Dosis del medicamento sospechoso (y, cuando sea relevante, otros datos tales como forma farmacéutica y vía de administración).
• Fecha de la reacción adversa.
• Fechas de administración del medicamento.
• Descripción de la reacción adversa.
• Resultado (por ej., resuelto, fatal, mejorado, secuela, desconocido).
Esto debe indicar las consecuencias de las reacciones adversas para el
paciente, utilizando el peor de los diferentes resultados para
múltiples reacciones.
2.6.8. Estudios
Todos los estudios (no clínicos, clínicos y epidemiológicos) que
contengan información de seguridad (incluyendo falta de eficacia) con
un potencial impacto en la información del producto, concluidos o en
progreso y los publicados que notifiquen situaciones de seguridad serán
incluidos en una discusión de los resultados finales o provisorios. Los
estudios que son parte del Plan de Gestión de Riesgo serán también
mencionados.
2.6.9. Otra información
2.6.9.1. Información relacionada a calidad
En esta sección se deberán informar y analizar todos los reportes de
falta de eficacia y pro-blemas/desvíos de calidad del producto.
2.6.9.2. Plan de Gestión de Riesgo
Debe informarse la eventual existencia de un Plan de Gestión de Riesgo y analizarse cualquier dato relevante surgido de aquél.
2.6.10. Evaluación Global de Seguridad
El TARC debe aportar un análisis conciso de todos los datos recopilados
durante el período en cuestión, incluyendo una evaluación de la
significación de dichos datos. El TARC debe, además, rever la
experiencia acumulada y resaltar cualquier nueva información con
respecto a:
• Un cambio en las características de las reacciones listadas, por ej., seriedad, resultado, población.
• Reacciones no listadas, serias y no serias.
• Una frecuencia incrementada de reporte de reacciones listadas.
• Interacciones.
• Experiencia con sobredosis, deliberada o accidental, y su tratamiento.
• Abuso o mal uso.
• Experiencias positivas o negativas durante el embarazo o lactancia.
• Experiencia en grupos especiales de pacientes.
• Efectos en el tratamiento a largo plazo.
• Errores de prescripción/de medicación.
2.6.11. Conclusión
La conclusión debe tratar el balance beneficio-riesgo en el contexto de
los datos presentados en el IPAS. También se indicarán qué datos de
seguridad no están en concordancia con la experiencia acumulada previa
y la información de referencia de seguridad. Se especificará y
justificará cualquier acción recomendada o iniciada. La necesidad de
modificar el prospecto será tratada cuando haya una inconsistencia
entre la información de referencia y el prospecto previo. Habiendo
tomado la decisión de modificar el prospecto, el TARC debe presentar la
solicitud al mismo tiempo que el IPAS o, si esto no es posible, indicar
una fecha para dicha solicitud.
3. PLAN DE GESTION DE RIESGO
3.1. Introducción
Un sistema de gestión de riesgos se define como un conjunto de
actividades e intervenciones en Farmacovigilancia diseñadas para
identificar, caracterizar, prevenir o minimizar riesgos relacionados
con productos medicinales, y la evaluación de la efectividad de esas
intervenciones. Este capítulo tiene como objetivo proveer una guía de
cómo un TARC debe presentar un Plan de Gestión de Riesgo (PGR) a la
Autoridad Regulatoria. La gestión de riesgo es un proceso continuo
durante todo el ciclo de vida de un producto medicinal y sus
actividades pueden cambiar por acontecimientos técnicos, científicos y
legislativos, así como también por la información disponible, los
riesgos percibidos y el impacto estimado en salud. Todos estos factores
deben tenerse en cuenta cuando se formula un PGR.
3.2. Descripción y requerimientos
3.2.1. Descripción del PGR
El objetivo de un PGR es asegurar que los beneficios de un medicamento
superen los riesgos por el mayor margen posible tanto para el paciente
como para la comunidad en su conjunto. Esto puede lograrse aumentando
los beneficios o disminuyendo los riesgos, pero, por definición, la
gestión de riesgos se ocupa de disminuir los riesgos probables.
3.2.2. Plan de Gestión de Riesgo (PGR)
Para la presentación de un PGR se requiere completar el formulario
correspondiente (ver Formulario 10 del Anexo II), cuya estructura se
ajusta a los siguientes lineamientos.
Un PGR contiene 2 partes:
Parte I:
• Especificaciones de seguridad.
• Plan de Farmacovigilancia.
Parte II:
• Evaluación de la necesidad de actividades de minimización de riesgos.
Y si hubiese la necesidad de actividades de minimización adicionales
(no rutina):
• Un plan de minimización de riesgos.
La Parte I incorpora los conceptos de la ICH (International Conference
on Harmonization) acerca de las especificaciones de seguridad que
resumen el perfil de seguridad del medicamento en el momento del ciclo
de vida en que se encuentre y el Plan de Farmacovigilancia basado en
dichas especificaciones. En la Parte II, basándose en las
especificaciones de seguridad, el TARC debe considerar la necesidad de
introducir medidas de minimización de riesgo. Estas actividades pueden
ser de rutina o adicionales (ver más adelante). Si se requieren sólo
actividades de rutina, no es necesaria la presentación de un plan de
minimización de riesgo. Si se planean actividades adicionales, el TARC
deberá presentar en la Parte II del PGR un plan de minimización de
riesgo que contenga tanto las actividades adicionales como las de
rutina. Es importante aclarar que el PGR debe presentarse siempre,
independientemente de si requerirá un plan de minimización de riesgo.
3.2.3. Situaciones en las que un PGR es necesario
Se debe presentar en las siguientes situaciones:
a) Para el Registro de la Especialidad Medicinal (REM), el TARC deberá presentar un PGR:
Con la solicitud de aprobación de:
• Un producto que contenga un nuevo ingrediente farmacéutico activo (nueva molécula).
• Un producto biológico, incluyendo a las vacunas.
• Un producto similar donde un problema de seguridad del producto de referencia requiere actividades de minimización de riesgos.
• Una asociación fija sin evidencia de comercialización en los países de Anexo I del Decreto 150/92.
• A pedido de la Autoridad Regulatoria Nacional.
• Un medicamento a registrarse bajo condiciones especiales (por ej., un medicamento huérfano).
b) Para modificaciones en el Registro de Especialidades Medicinales (REM), el TARC deberá presentar un PGR:
• Con la solicitud de aprobación de cambios importantes en la
especialidad medicinal: nueva dosis, nueva forma farmacéutica, nueva
vía de administración, cambios en la indicación, nuevo proceso de
fabricación de un producto biológico, etc., que implique un posible
riesgo.
• A pedido de la Autoridad Regulatoria Nacional.
c) Por iniciativa del TARC, al identificar un problema de seguridad con el medicamento en cualquier estadio de su ciclo de vida.
d) A pedido de la Autoridad Regulatoria Nacional (tanto pre como post autorización).
En cualquier otra situación que no sea obligatoria, el TARC y la
Autoridad Nacional deberán analizar la necesidad de crear un PGR.
3.3. Especificaciones de Seguridad
Deben ser un resumen de los riesgos identificados de un producto
medicinal, riesgos potenciales e información faltante. También deben
incluir poblaciones potencialmente en riesgo. Las especificaciones de
seguridad deben ayudar a la industria farmacéutica y a la autoridad
regulatoria a identificar cualquier necesidad de recolección especial
de datos y también facilitar la construcción del Plan de
Farmacovigilancia. El PGR también fundará las bases para la evaluación
de la necesidad de actividades de minimización de riesgo, y cuando sea
apropiado, el plan de minimización de riesgos. Se recomienda a los TARC
que sigan la estructura que se proveerá en estos ítems, aunque pueden
incluir elementos adicionales, dependiendo de la naturaleza del
producto y su programa de desarrollo. Por otra parte, para productos
que ya se encuentran en el mercado, y con nuevas cuestiones de
seguridad, solamente deberán presentar algunos elementos.
3.3.1. Pre-clínico
Dentro de las especificaciones de seguridad, esta sección debe
presentar datos no clínicos que no hayan sido adecuadamente evaluados
con datos clínicos, tales como:
• Toxicidad (neurotoxicidad, genotoxicidad, etc.)
• Farmacología general (cardiovascular, incluyendo prolongación del QT, sistema nervioso, etc.)
• Interacciones de drogas.
• Otra información de toxicidad.
3.3.2. Clínico
3.3.2.1. Limitaciones de la base de datos de seguridad
Serán consideradas las limitaciones de la base de datos (por ej.,
relacionado al tamaño de la población en estudio, criterios de
inclusión y exclusión de los estudios). Se deben discutir las
implicaciones de dichas limitaciones con respecto a la predicción de
seguridad de la especialidad medicinal en el mercado. Se debe hacer
particular referencia a poblaciones que puedan ser expuestas durante el
uso del producto en la práctica médica.
Exposición Post comercialización
Cuando la droga esté comercializada, el TARC debe proveer los datos de
los pacientes ya expuestos. Cuando se decida qué medida utilizar para
medir la exposición es importante considerar cómo se utiliza el
medicamento. Por ej., en medicamentos utilizados en uso crónico, la
medida apropiada podría ser “paciente años de uso”. Sin embargo, cuando
el uso es limitado y generalmente determinado por cantidad, por ej.,
antibióticos, una suma de los envases vendidos puede ser suficiente. La
información debe ser estratificada para variables significativas, como
edad, indicación, dosis y duración de tratamiento.
3.3.2.2. Poblaciones no estudiadas en fases pre-autorización
La especificación de seguridad debe discutir cuáles son las poblaciones
que no han sido estudiadas o han sido poco estudiadas. Las poblaciones
consideradas para discusión deben incluir (no limitarse únicamente a
las siguientes):
• Niños.
• Ancianos.
• Embarazadas o mujeres lactando.
• Pacientes con co-morbilidad relevante, como desórdenes hepáticos o renales.
• Pacientes con enfermedades severas distintas a las estudiadas en los ensayos clínicos.
• Poblaciones que presenten polimorfismos genéticos relevantes.
• Pacientes con diferentes orígenes raciales o étnicos.
3.3.2.3. Reacciones Adversas
Esta sección debe listar los riesgos identificados importantes y riesgos potenciales que requieren mayor análisis o evaluación.
• Riesgos identificados que requieren mayor evaluación
Será incluida información detallada de las reacciones adversas
identificadas como más importantes, que incluirá aquellas serias o
frecuentes y que además presenten un impacto en el balance
riesgo-beneficio. La información incluirá la evidencia basada en
relación causal, seriedad, frecuencia, reversibilidad y grupos de
riesgo, si hay disponible. También se discutirán los factores de riesgo
y mecanismos potenciales. Estas reacciones adversas usualmente incluyen
mayor información de evaluación en el Plan de Farmacovigilancia.
• Riesgos potenciales que requieren mayor evaluación
En esta sección se describirán los riesgos potenciales importantes. Se
presentará la evidencia que llevó a la conclusión que se trata de un
riesgo. Se anticipa que ante cualquier potencial riesgo, debe haber
mayor evaluación que caracterice a la asociación.
3.3.2.4. Interacciones potenciales e identificadas incluyendo alimentos-medicamentos y fitoterápicos-medicamentos
Se discutirán las interacciones farmacocinéticas y farmacodinámicas
potenciales e identificadas. Para cada una, se resumirá la evidencia
que incluye sus posibles mecanismos y los potenciales riesgos en salud
para las distintas indicaciones y distintas poblaciones. Se debe
aclarar cuáles de las interacciones requieren mayor evaluación.
3.3.2.5. Epidemiología
La epidemiología de las indicaciones debe ser discutida, y debe incluir
incidencia, prevalencia, mortalidad y co-morbilidades relevantes. De
estar disponible, se incluirá también la información acerca de factores
de riesgo.
3.3.2.6. Efectos de clase farmacológicos
Las especificaciones de seguridad deben identificar riesgos que, se
cree, se relacionan a la clase farmacológica. Si un riesgo, que sea
comúnmente relacionado a la clase, no pareciera ser una cuestión de
seguridad, esto se deberá justificar.
3.3.2.7. Otros requerimientos
El TARC debe discutir los siguientes ítems. Si se cree que el riesgo
potencial es significativo, el ítem será identificado como un riesgo
importante y las maneras para reducirlo o minimizarlo serán discutidas
en “evaluación de la necesidad de actividades de minimización de
riesgo”. En este contexto “significativo” significa que hay una
posibilidad razonable de que ocurra.
• Sobredosis potencial (por ej., margen de seguridad estrecho).
• Potencial transmisión de agentes infecciosos.
• Potencial uso ilegal.
• Potencial uso fuera de prospecto (off-label).
• Potencial uso fuera de prospecto en poblaciones pediátricas.
3.3.3. Resumen
Al final de las especificaciones de seguridad se incluirá un resumen acerca de:
• Riesgos identificados importantes.
• Riesgos potenciales importantes.
• Información faltante importante.
Basado en este resumen, el TARC proveerá un Plan de Farmacovigilancia y
la evaluación de la necesidad de actividades de minimización de riesgos.
3.4. Plan de Farmacovigilancia
El Plan de Farmacovigilancia debe basarse en las especificaciones de
seguridad y debe proponer acciones para los problemas de seguridad
identificados. Es importante aclarar que sólo una proporción de los
riesgos son previamente identificados y el Plan de Farmacovigilancia no
reemplaza, sino que complementa los procedimientos actuales para la
detección de señales.
3.4.1. Farmacovigilancia de rutina
Para productos que no presenten riesgos especiales, la
Farmacovigilancia de rutina debe ser suficiente para el monitoreo post
comercialización, sin la necesidad de acciones complementarias (por
ej., estudios de seguridad).
3.4.2. Planes de acción y actividades adicionales de Farmacovigilancia
Para productos medicinales con riesgos importantes identificados,
riesgos potenciales y/o información faltante importante, se planearán
actividades adicionales para esos problemas de seguridad. Los objetivos
variarán de acuerdo al problema de seguridad. Por ej., si falta
información importante, el objetivo puede ser reafirmar la ausencia del
riesgo.
3.4.3. Planes de acción para problemas de seguridad
Dentro del Plan de Farmacovigilancia, el plan de acción para cada
problema de seguridad debe ser presentado y justificado acorde a la
siguiente estructura:
• Problema de seguridad.
• Objetivo de las acciones propuestas.
• Acciones propuestas.
• Justificación para las acciones propuestas.
• Monitoreo por el TARC.
• Metas para la evaluación y el reporte.
3.5. Evaluación de las necesidades de actividades de minimización de riesgo
Para cada problema de seguridad, el TARC debe evaluar la necesidad de
actividades de minimización de riesgo. Algunas situaciones de seguridad
podrán ser resueltas con las acciones propuestas en el Plan de
Farmacovigilancia, pero para otras, el riesgo puede ser de una
naturaleza particular y de seriedad tal que requieran actividades de
minimización de riesgos. Es posible que estas actividades estén
limitadas a asegurar que las precauciones adecuadas estén incluidas en
los prospectos. Sin embargo, para algunos riesgos, las actividades de
rutina no son suficientes. De ser requeridas, estas actividades deben
ser descriptas en el Plan de Minimización de Riesgos.
3.6. Plan de Minimización de Riesgos
El Plan de Minimización de Riesgos detalla las actividades de
minimización de riesgo que se llevarán a cabo para reducir los riesgos
asociados a un problema particular de seguridad. Dicho plan debe
incluir las actividades tanto de rutina como adicionales. Un problema
de seguridad puede incluir más de una actividad de minimización de
riesgo. Un ejemplo es un posible plan para un agente teratógeno, que
puede tener como objetivo evitar la toma de medicación en mujeres
embarazadas. Una actividad de rutina incluiría agregar en el prospecto
una advertencia al respecto y solicitar que las mujeres presenten un
test de embarazo negativo antes de empezar el tratamiento. Actividades
adicionales pueden incluir un folleto educativo dirigido a pacientes
sobre los riesgos del medicamento y la necesidad de la anticoncepción.
Por cada propuesta se debe incluir cómo se va a medir la efectividad de
cada acción.
3.6.1. Errores de medicación potenciales
El TARC debe considerar sistemáticamente la posibilidad de que se
cometan errores de medicación teniendo en cuenta las fuentes de error
de medicación más comunes y debe analizar las acciones a tomar para
evitarlos.
3.7. Actividades de minimización de riesgo
Es dificultoso proveer una guía precisa acerca de qué actividad debe
ser utilizada en cada situación, debiéndose considerar las necesidades
de seguridad caso por caso. Es esencial consultar con los expertos
especializados en todos los estadios.
3.7.1. Comunicación del riesgo
La comunicación precisa y acorde de datos emergentes en seguridad es
una parte esencial de la farmacovigilancia. Los pacientes y los
profesionales de seguridad requieren una información segura y bien
comunicada acerca de los riesgos asociados al producto medicinal y la
condición por la cual se utiliza. Debido a la importancia de la
comunicación del riesgo se recomienda que sean consultados expertos en
el área.
3.8. Asegurar la efectividad de las actividades de minimización de riesgo
La definición de gestión de riesgo requiere evaluación de la
efectividad de las intervenciones que forman parte del proceso. La
evaluación de la efectividad incrementará el entendimiento de qué
actividades son las más apropiadas según el tipo de cuestión de
seguridad.
3.8.1. Evaluación de la minimización del riesgo
Se emplearán mediciones directas de minimización de riesgo en caso de
estar disponibles. Se considerarán medidas secundarias cuando esto no
sea posible o para proveer asesoramiento interno, mientras se espera el
resultado de las mediciones directas. Por ej., mediciones basadas en la
provisión de información a profesionales, estudios descriptivos o
encuestas que evalúen si la información es comunicada efectivamente.
3.9. Resumen de las actividades en el PGR
El PGR debe contener un resumen completo de las actividades detalladas
para el producto medicinal. Debe ser realizado en dos partes:
• Resumen de todas las actividades (Farmacovigilancia y minimización de
riesgo) para cada cuestión importante de seguridad. Debe ser una tabla
que enumere cada cuestión de seguridad.
• Resumen de todas las actividades y sus objetivos. Esta sección debe
ser organizada en forma tal que las actividades llevadas a cabo
coincidan con sus objetivos. La razón para esto es que una actividad
propuesta puede tratar más de una cuestión de seguridad. Líneas de
tiempo y objetivos también se incluyen en el resumen.
3.10. Presentación de documentos actualizados
La presentación del documento actualizado debe incluir resultados de la
evaluación periódica de la efectividad de las actividades del PGR. Si
surge información adicional de seguridad del producto ésta se incluirá
en una nueva versión de PGR y deberá considerarse si nuevas actividades
de minimización de riesgo son necesarias. La actualización deberá
presentarse:
• Dentro los 6 meses o el año, según la especialidad medicinal, de la
ejecución del Plan de Farmacovigilancia o de las actividades de
minimización de riesgo o cuando resultados de estudios se encuentren
disponibles.
• En caso que una nueva información recibida impacte en las
especificaciones de seguridad en el Plan de Farmacovigilancia o en las
actividades de minimización de riesgo.
• Cuando lo requiera la Autoridad Regulatoria Nacional.
4. BUENAS PRACTICAS DE FARMACOVIGILANCIA EN VACUNAS
4.1. Introducción
La Farmacovigilancia es la ciencia y las actividades relacionadas con
la detección, evaluación, comprensión y prevención de reacciones
adversas y otros posibles problemas relacionados con los medicamentos.
Recientemente, las incumbencias de la Farmacovigilancia han sido
extendidas, incluyendo: hierbas, medicamentos tradicionales y
complementarios, productos hemoderivados, biológicos y vacunas. Las
vacunas son productos inmunobiológicos o agentes inmunizantes que se
utilizan para la producción de respuestas inmunitarias específicas
protectoras (anticuerpos y/o inmunidad mediada por células). Aunque su
descubrimiento y su introducción comenzaron a fines del siglo XVIII, su
potencial no fue reconocido hasta erradicar la viruela. La prevención
de enfermedades a través de las vacunas ha sido uno de los mayores
logros para la Salud Pública.
Ningún medicamento es cien por ciento seguro y cien por ciento eficaz y
las vacunas no escapan a esta regla; es por esto que se debe realizar
un monitoreo de los eventos supuestamente atribuibles a vacunación e
inmunización (ESAVI) y seguir lineamientos que se desarrollarán en las
Buenas Prácticas de Farmacovigilancia en Vacunas. Estas Buenas
Prácticas están destinadas a evaluar en forma correcta los eventos
asociados a vacunas y vacunación, utilizando criterios uniformes en la
evaluación de los ESAVI y en la generación de señales y alertas. El
Sistema Nacional de Farmacovigilancia ha incluido desde el comienzo
(1993) el seguimiento de los eventos adversos a vacunas dentro de sus
responsabilidades, acorde a las definiciones de la OMS del año 2002.
4.2. Eventos supuestamente atribuibles a vacunación e inmunización (ESAVI)
4.2.1. Concepto de ESAVI
Es un cuadro clínico que tiene lugar después de la administración de
una vacuna, que podría o no estar relacionado con ésta y que causa gran
preocupación en la población.
4.2.2. Notificación de ESAVI
La buena documentación constituye parte fundamental de un sistema de
garantía de calidad y de las Buenas Prácticas de Farmacovigilancia. La
importancia de esto radica en que lo notificado puede generar señales.
Por ello, la calidad de las notificaciones es crítica para una
apropiada evaluación de la relación entre la aplicación de la vacuna y
la aparición del ESAVI. La principal fuente de información en
Farmacovigilancia es la notificación espontánea de eventos
supuestamente atribuibles a vacunaciones o inmunizaciones. Tan
importantes como la notificación espontánea, son los métodos de
Farmacovigilancia activa, que proporcionan datos relevantes y
específicos en poblaciones especiales y vacunas nuevas. Las
notificaciones de eventos adversos del Sistema Nacional de
Farmacovigilancia se caracterizan por ser voluntarias, espontáneas y
confidenciales. Son especialmente útiles en detectar señales de eventos
adversos raros, serios o inesperados. Se efectúan en un formulario de
notificación para ESAVI (Ver Formulario 2 Anexo II). Es indispensable
que los reportes enviados tengan como información mínima: la edad del
paciente, la fecha de aplicación de la vacuna, la fecha del ESAVI,
vacunas aplicadas en forma concomitante, marca comercial y número de
lote de la vacuna sospechosa.
4.2.3. Clasificación de los ESAVI
1) Eventos no relacionados o coincidentes
Se trata de eventos que ocurren después de la vacunación, pero que no
son causados por las vacunas: es una asociación azarosa, es decir,
existe una relación temporal pero no de causa-efecto (son eventos
independientes).
2) Eventos relacionados
2a) Eventos relacionados con problemas operativos del programa (Error
Programático). Es un evento causado en el ciclo de uso de la vacuna por
un error en su almacenamiento, preparación y manejo o administración.
Los eventos causados por error programático, es decir, error operativo
del programa, son prevenibles por el vacunador. Por ejemplo, aplicar
inyecciones no estériles, errores en la reconstitución, inyecciones en
el lugar equivocado, administración por una vía diferente a la
establecida, transporte o almacenamiento incorrecto de las vacunas,
inobservancia de las contraindicaciones.
2b) Eventos relacionados con la vacuna. La vacuna se aplicó
correctamente pero, debido a sus propiedades o componentes, causó el
evento adverso o lo precipitó. Los eventos relacionados con la vacuna
son habitualmente no serios.
* Reacciones intrínsecas: respuesta del organismo asociada al producto biológico propiamente dicho.
* Reacciones extrínsecas: frente a una reacción vacunal hay que tener
en cuenta que otros componentes de la formulación podrían causar los
eventos observados (eventos extrínsecos) y que muchas veces las
reacciones varían en intensidad y forma; se las asocia equivocadamente
con el producto biológico de la vacuna, pero son reacciones del
organismo a los coadyuvantes de la formulación (por ejemplo agentes de
reconstitución, agentes preservantes, estabilizantes, adyuvantes,
antibióticos, etc.)
* Factores relacionados con el paciente: evento causado por
susceptibilidad genética, ansiedad o dolor por la inyección en sí misma
y no por la vacuna.
* Desvío de calidad: es el distanciamiento de los parámetros aptos para
la aprobación de la vacuna, por ejemplo, el aumento de la concentración
viral.
3) La investigación no es concluyente
Cuando no es posible determinar la relación de causalidad.
4.3. Responsabilidades ANMAT - Farmacovigilancia
- Investigar, procesar, codificar, clasificar, digitalizar (base de datos) y analizar el riesgo y la relevancia de los ESAVI.
- Proponer acciones sanitarias o regulatorias.
- Enviar los datos al Centro Colaborativo de la Organización Mundial de la Salud (UMC) a través de su base de datos Vigiflow.
El Departamento de Farmacovigilancia recibe las notificaciones de eventos supuestamente causados por:
a) Vacunas que figuran en el Calendario Nacional de Vacunación: se
comparte la investigación con el Programa de Enfermedades
Inmunoprevenibles del Ministerio de Salud de Nación.
b) Vacunas fuera de calendario: la investigación de los casos de ESAVI
se realiza desde el Departamento de Farmacovigilancia de ANMAT con
colaboración del Instituto Nacional de Medicamentos (INAME) y del
laboratorio productor.
4.4. Investigación de los eventos supuestamente atribuibles a la vacunación e inmunización (ESAVI)
Todo evento adverso que los pacientes o los profesionales de la salud
consideren relacionado con una vacuna debe investigarse. Si el período
y los síntomas indican la posibilidad de que ese evento tenga una
relación con la vacuna, se iniciará de inmediato una investigación. La
finalidad de la investigación es confirmar o descartar el evento
notificado, determinar si existen otras causas posibles, verificar si
se trata de un hecho aislado o en un grupo de personas e informar a las
partes involucradas.
4.4.1. Aspectos clave de los ESAVI
• No existe una vacuna ideal que proteja a todos los vacunados y que sea absolutamente segura.
• Las vacunas eficaces (es decir, que inducen inmunidad protectora)
pueden producir algunos efectos secundarios no deseables que son, por
lo general, no serios y desaparecen rápidamente.
• No es posible predecir en todos los casos quienes pueden sufrir reacciones serias o no serias a una vacuna.
• El tamaño de los lotes de vacunas puede oscilar desde varios cientos
de miles a varios millones de dosis y algunos lotes de vacunas se
distribuyen durante períodos mucho más largos que otros. Naturalmente,
un lote de mayor tamaño o uno que se distribuye durante un período más
largo estará asociado, simplemente por efecto del azar, con un número
mayor de incidentes adversos. Además, dado que las tasas de mortalidad
infantil son máximas durante el primer año de vida, se asocia un mayor
número de fallecimientos producidos de forma coincidente con las
vacunas administradas a lactantes que con las vacunas proporcionadas en
momentos posteriores de la infancia.
4.5. Responsabilidades del Titular de Registro y Autorización de Comercialización (TARC)
• Realizará la vigilancia post-comercialización de las vacunas que produce.
• Mantendrá informado al Departamento de Farmacovigilancia de la ANMAT
de cualquier evento adverso que sea notificado localmente.
• Procesará los datos obtenidos y los difundirá en un informe de
actualización de seguridad semejante al que se presenta con
medicamentos.
• Mantendrá una comunicación permanente con el Departamento de Farmacovigilancia de la ANMAT.
• Enviará al Sistema Nacional de Farmacovigilancia de la ANMAT la
información sobre la vigilancia de eventos adversos según la
periodicidad establecida, serios en 72 hs. desde su conocimiento y los
no serios bimestralmente.
• Participar de la investigación de los eventos adversos serios cuando se los convoque o solicite información.
• Es recomendable que el responsable de los eventos adversos de vacunas
sea un personal especializado, por ej., un médico infectólogo.
ANEXO II
FORMULARIOS
1. Comunicación de eventos adversos
2. Comunicación de eventos adversos supuestamente atribuibles a la vacunación e inmunización (ESAVI)
3. Comunicación de desvíos de calidad
4. Comunicación de eventos adversos por uso de medicamentos
fitoterápicos, productos vegetales y/o preparados de drogas vegetales
5. Comunicación de errores de medicación
6. Comunicación de eventos adversos. Formulario para pacientes
7. Comunicación eventos adversos en formato CIOMS
8. Comunicación de reportes bimestrales de eventos adversos no serios para la industria farmacéutica
9. Comunicación de reportes bimestrales por falta de eficacia/desvío de calidad para la industria farmacéutica
10. Presentación de los Planes de Gestión de Riesgo (PGR)
INSTRUCCIONES PARA NOTIFICACIONES DE EVENTOS ADVERSOS
Lea atentamente toda la hoja antes de completarla, de este modo podrá
llenarla con la mayor cantidad de datos posibles en sus casilleros
correspondientes.
Escriba con letra clara, esto facilitará la evaluación y clasificación del evento.
Se define evento adverso como cualquier suceso médico nocivo y no
intencionado que puede presentarse durante el tratamiento con un
producto, pero que no tiene necesariamente una relación causal con el
mismo. Un efecto adverso es una reacción nociva y no deseada que se
presenta tras la administración de un medicamento, a dosis utilizadas
habitualmente en la especie humana, para prevenir, diagnosticar o
tratar una enfermedad, o para modificar cualquier función biológica.
Datos del paciente: Escriba los datos conocidos del paciente. Puede usar iniciales para proteger la identidad del mismo.
Descripción del evento adverso: Indique los signos y síntomas del
evento adverso que desencadenó la notificación, incluyendo fecha de
inicio y finalización. Aunque se trate de una reacción adversa
conocida, es importante su notificación.
Medicamento: Escriba en primer lugar el producto que usted cree que es
el responsable del evento adverso. Indique el nombre genérico y el
nombre comercial; la dosis; su frecuencia y vía de administración; las
fechas de comienzo y final del tratamiento; las indicaciones de uso;
número de dosis recibidas por el paciente; si se sospecha de falta de
eficacia es importante consignar el nombre comercial, la fecha de
vencimiento y el número de lote.
Exámenes complementarios relevantes: Describa si existen exámenes
complementarios de importancia que sean relevantes en el evento adverso
junto con su resultado.
Condiciones médicas relevantes: Indique la enfermedad de base y toda condición médica previa, de importancia.
Medicación concomitante: Indique si el paciente recibió otra medicación
o terapias alternativas (hierbas, venenos de serpientes, medicamentos
homeopáticos, etcétera).
Resultado: Marque con una cruz los casilleros necesarios.
Datos del comunicador del evento adverso (optativo): Pueden ser sólo
iniciales y contar con lo indispensable para canalizar una respuesta,
si fuera necesario.
INSTRUCCIONES PARA NOTIFICACIONES DE EVENTOS ADVERSOS SUPUESTAMENTE ATRIBUIBLES A LA VACUNACION E INMUNIZACION (ESAVI)
Lea atentamente toda la hoja antes de completarla, de este modo podrá
llenarla con la mayor cantidad de datos posibles en sus casilleros
correspondientes.
Escriba con letra clara, esto facilitará la evaluación y clasificación del evento.
Tipo de ESAVI: Un ESAVI es un cuadro clínico que tiene lugar después de
la administración de una vacuna, que podría o no estar relacionado con
ésta. Marque con una cruz si sospecha un evento asociado a la vacuna o
bien asociado a la práctica vacunatoria.
Datos del paciente: Escriba los datos conocidos del paciente. Puede
usar iniciales para proteger la identidad del mismo. Peso y talla son
muy necesarios en el caso de notificaciones pediátricas.
Descripción del evento adverso: Indique los signos y síntomas del
evento adverso que desencadenó la notificación, incluyendo fecha de
inicio y finalización. Aunque se trate de una reacción adversa
conocida, es importante su notificación.
Vacuna: Escriba en primer lugar la vacuna que usted cree que es la
responsable del evento adverso. Indique el tipo de vacuna; la dosis;
sitio de aplicación; fecha de vacunación; laboratorio y productor;
número de lote y serie. Indique si el paciente recibió en forma
concomitante otras vacunas y especifíquelas.
Exámenes complementarios relevantes: Se deberán indicar todos los
estudios que se hayan realizado durante el episodio y la evolución del
presunto ESAVI, junto con su resultado. Ej: Laboratorios específicos
y/o rutinas, radiografías, ECG, EEG, etc.
Condiciones médicas relevantes: Marque con una cruz toda condición médica previa que tenga el paciente.
Medicación concomitante: Indique si el paciente recibió otra medicación
o terapias alternativas (hierbas, venenos de serpientes, medicamentos
homeopáticos, etcétera).
Resultado del ESAVI: Marque con una cruz los casilleros necesarios.
Lugar de vacunación: Marque con una cruz el lugar físico donde fue aplicada la vacuna.
Marco de aplicación de la vacuna: Marque con una cruz el motivo por el cual fue aplicada la vacuna.
Datos del comunicador del evento adverso (optativo): Pueden ser sólo
iniciales y contar con lo indispensable para canalizar una respuesta,
si fuera necesario.
INSTRUCCIONES PARA NOTIFICACIONES DE DESVIOS DE CALIDAD
Lea atentamente toda la hoja antes de completarla, de este modo podrá
llenarla con la mayor cantidad de datos posibles en sus casilleros
correspondientes.
Escriba con letra clara, esto facilitará la evaluación y clasificación del evento.
Desvío de calidad:
Se define como desvío de calidad a fallas en la calidad producidas
durante el proceso de elaboración y que son responsabilidad del
laboratorio productor.
Datos del paciente: Escriba los datos conocidos del paciente. Puede usar iniciales para proteger la identidad del mismo.
Descripción del desvío de calidad: Indique las características del
desvío, aclarando si el mismo causó un evento adverso en el paciente
(reacción adversa ligada a la calidad). Se define falta de efectividad
cuando un producto no produce la respuesta terapéutica esperada (de
acuerdo a los antecedentes del fármaco y a las condiciones del
paciente). Es necesario especificar cuál es el efecto terapéutico o
farmacológico que no ha sido percibido. Considere la posibilidad de
individuos no respondedores, cambio del producto por otro nombre
comercial, falta de cumplimiento del tratamiento, deficiencia en la
calidad farmacéutica del producto, etcétera. En el caso de otros
desvíos de calidad, es necesario especificar la cantidad de unidades
del producto que tienen el desvío (Ej.: de una caja de 100 ampollas, 4
ampollas con partículas visibles).
Medicamento: En cuanto a los medicamentos sospechosos, considere tanto
los principios activos como los excipientes. Indique el nombre genérico
y el nombre comercial; la dosis; vía de administración; forma
farmacéutica; indicaciones de uso; número de dosis recibidas por el
paciente; la fecha de vencimiento y el número de lote.
Envío de muestras: Enviar la muestra en su envase original, sin abrir.
Las muestras deberán ser enviadas respetando las condiciones de
almacenamiento mencionadas en el envase y/o prospecto. Si la muestra
remitida no se envía en las condiciones que el producto requiere, la
misma no será procesada por el SNFVG. Tampoco se procesarán los
productos vencidos. En el caso de desvíos de rótulos, deberá enviarse
un envase del producto (que puede estar abierto) o bien la fotocopia de
todas las caras del envase donde se observa el error.
| Muestras que no se deben enviar a FVG |
| Magistrales | Homeopáticos | Alimentos | Suplementos dietarios | Productos médicos | Cosméticos | Ilegítimos o falsificados | Reactivos de diagnóstico |
| Contacto | Colegio farmacéutico de cada región | Vigilancia alimentaria 4340-0800 Int. 3537 4340-0888 vigi.alimentaria@anmat.gov.ar | Tecnovigilancia 4340-0800 Int. 1511 | Cosmetovigilancia 4340-0800 Int 2573 | Programa Nacional de Control de Mercado 4340-0800 Int. 2562 | Servicio de Productos para Diagnóstico 4340-0800 Int. 2704 |
Exámenes complementarios relevantes: Describa si existen exámenes
complementarios de importancia que sean relevantes en el evento adverso
junto con su resultado.
Condiciones médicas relevantes: Indique la enfermedad de base y toda condición médica previa, de importancia.
Medicación concomitante: Indique si el paciente recibió otra medicación
o terapias alternativas (hierbas, venenos de serpientes, medicamentos
homeopáticos, etcétera).
Resultado: Marque con una cruz los casilleros necesarios.
Datos del comunicador del evento adverso (optativo): Pueden ser sólo
iniciales y contar con lo indispensable para canalizar una respuesta,
si fuera necesario.
INSTRUCCIONES PARA NOTIFICACIONES DE EVENTOS ADVERSOS POR USO DE
MEDICAMENTOS FITOTERAPICOS, PRODUCTOS VEGETALES Y/O PREPARADOS DE
DROGAS VEGETALES
Lea atentamente toda la hoja antes de completarla, de este modo podrá
llenarla con la mayor cantidad de datos posibles en sus casilleros
correspondientes.
Escriba con letra clara, esto facilitará la evaluación y clasificación del evento.
Tipo de Evento Adverso:
Marque con una cruz si sospecha un evento adverso farmacológico, un
efecto tóxico o una falta de efectividad del producto. Se define evento
adverso como cualquier suceso médico nocivo y no intencionado que puede
presentarse durante el tratamiento con un producto, pero que no tiene
necesariamente una relación causal con el mismo. Se define falta de
efectividad cuando un producto no produce la respuesta terapéutica
esperada (de acuerdo a los antecedentes del mismo y a las condiciones
del paciente). Considere la posibilidad de individuos no respondedores,
cambio del producto por otro nombre comercial, falta de cumplimiento
del tratamiento, deficiencia en la calidad farmacéutica del producto,
etcétera. En caso de sospecha de falta de efectividad trate de
completar el casillero de nombre comercial, fecha de vencimiento y
número de lote.
Datos del paciente: Escriba los datos conocidos del paciente. Puede usar iniciales para proteger la identidad del mismo.
Descripción del evento adverso: Indique los signos y síntomas del
evento adverso que desencadenó la notificación, incluyendo fecha de
inicio y finalización. Aunque se trate de una reacción adversa
conocida, es importante su notificación.
Medicamentos y/o productos vegetales: Escriba en primer lugar el
producto que usted cree que es el responsable del evento adverso.
Indique el nombre común y el nombre comercial; la dosis; su frecuencia
y vía de administración; las fechas de comienzo y final del
tratamiento; las indicaciones de uso. En caso de productos vegetales,
indicar parte usada de la planta y forma de preparación (té, infusión,
etc.). Indique la forma de obtención del material vegetal, recolectado
(obtenido de su ambiente natural) o adquirido, en farmacias,
herboristerías, etcétera.
Exámenes complementarios relevantes: Describa si existen exámenes
complementarios de importancia que sean relevantes en el evento adverso
junto con su resultado.
Condiciones médicas relevantes: Indique la enfermedad de base y toda condición médica previa, de importancia.
Medicación concomitante: Indique si el paciente recibió otra medicación
o terapias alternativas (hierbas, venenos de serpientes, medicamentos
homeopáticos, etcétera).
Resultado: Marque con una cruz los casilleros necesarios.
Datos del comunicador del evento adverso (optativo): Pueden ser sólo
iniciales y contar con lo indispensable para canalizar una respuesta,
si fuera necesario.
Formulario para la presentación de los Planes de Gestión de Riesgo (PGR)
Para la presentación de los PGR se requiere completar el siguiente
formulario con los datos que se encuentren disponibles para la
especialidad medicinal. Esta estructura sigue los lineamientos para PGR
de las Buenas Prácticas de Farmacovigilancia. La información presentada
dependerá de la especialidad medicinal y la etapa del ciclo de vida en
que se encuentre la misma. Con excepción de la sección 4, anexos 2 y 3
(que deben ser completados en el caso de que se realicen esas acciones
particulares) todas las secciones deben ser presentadas.
Indice: Secciones
Información del producto
1. Especificaciones de Seguridad
2. Plan de Farmacovigilancia
3. Evaluación de la necesidad de las acciones de minimización de riesgo
4. Plan de minimización de riesgo
5. Resumen del PGR
6. Profesional de contacto con farmacovigilancia
7. Presentación de documentos actualizados
Anexo 1 Resumen de los protocolos de estudios de fase IV propuestos o en curso en Argentina
Anexo 2 Detalles del plan de minimización riesgo
Anexo 3 Rótulos y prospecto actuales o propuestos (en el caso de solicitud de registro)
Información del producto
| Nombre comercial |
|
| Ingrediente(s) farmacéutico(s) activo(s) (IFA) |
|
| Grupo farmacoterapéutico (Código ATC) |
|
| Nombre del Titular de Autorización de Registro (TAR) |
|
| Fecha y primer país de lanzamiento |
|
| Países en los que se comercializa actualmente |
|
| Fecha de presentación del PGR |
|
| Versión N° |
|
| Breve descripción del producto (clase química, mecanismo de acción, etc.) |
|
| Indicación(es) (aprobada y/o propuesta) |
|
| Posología (aprobada y/o propuesta) | Para cada indicación y duración de la terapia |
| Forma farmacéutica y concentración(es) |
|
1. Especificaciones de Seguridad del producto
1.1. Preclínico
1.1.1. Resumen de los hallazgos de seguridad preclínicos
Toxicidad (incluyendo la toxicidad de dosis repetidas, toxicidad
reproductiva/desarrollo, nefrotoxicidad, hepatotoxicidad,
genotoxicidad, carcinogenicidad)
Hallazgos generales de seguridad farmacológica (cardiovascular
[incluyendo la prolongación del intervalo QT], el sistema nervioso,
etc.)
Mecanismos de interacción con otros medicamentos
Otra información o datos relacionados con la toxicidad
| Problema de seguridad (a partir de los estudios pre-clínicos) | Relevancia para el uso humano |
| Toxicidad a dosis repetidas |
|
| Toxicidad
reproductiva [cuando el medicamento está diseñado para el uso en
mujeres en edad fértil se debe incluir un resumen de hallazgos
importantes (incluyendo negativos)] |
|
| Toxicidad del desarrollo |
|
| Etc. |
|
1.2. Clínico
1.2.1. Limitaciones de la base de datos de seguridad
1.2.1.1. Exposición en ensayos clínicos
Serán consideradas las limitaciones de la base de datos (por ej.,
relacionado al tamaño de la población en estudio, criterios de
inclusión y exclusión de los estudios) y se deben discutir las
implicaciones de dichas limitaciones con respecto a la predicción de
seguridad de la especialidad medicinal en el mercado. Se debe hacer
particular referencia a poblaciones que puedan ser expuestas durante el
uso del producto en la práctica médica.
1.2.1.2. Exposición poscomercialización
Los datos de los pacientes expuestos durante la comercialización se
deben obtener siempre que sea posible sobre la base de estudios de
mercado y se debe justificar la manera de realizar el cálculo de la
exposición.
1.2.2. Poblaciones no estudiadas en fases pre-autorización
La especificación de seguridad debe discutir cuáles son las poblaciones
que no han sido estudiadas o han sido poco estudiadas. Las poblaciones
consideradas para discusión deben incluir (no limitarse únicamente a
las siguientes):
• Niños.
• Ancianos.
• Embarazadas o mujeres lactando.
• Pacientes con co-morbilidad relevante, como desórdenes hepáticos o renales.
• Pacientes con enfermedades severas distintas a las estudiadas en los ensayos clínicos.
• Poblaciones que presenten polimorfismos genéticos relevantes.
• Pacientes con diferentes orígenes raciales o étnicos.
1.2.3. Reacciones adversas
1.2.3.1. Nueva información de seguridad desde la última presentación del PGR (para los documentos actualizados)
| Tema de seguridad 1 |
| Descripción |
| Fuente de la evidencia |
| ¿Se proponen nuevos estudios para el plan de farmacovigilancia Sí/No |
| ¿Se proponen nuevas acciones de minimización de riesgo Sí/No |
| Tema de seguridad 2 |
1.2.3.2. Descripción de los riesgos identificados y potenciales importantes
Para cada riesgo identificado y potencial importante presentar la siguiente información si está disponible:
| Riesgo Identificado/Potencial | Especificar el riesgo |
| Frecuencia y seriedad del riesgo |
|
| Frecuencia de los resultados | Calcular
la distribución de los resultados por ejemplo, % muerte, % se recuperó
con/sin tratamiento/secuelas, % no se recuperó, % hospitalizados |
| Antecedentes de incidencia/prevalencia en la población objetivo |
|
| Factores/grupos de riesgo | Describir el uso, dosis, tiempo y datos de susceptibilidad o de otros factores que estén disponibles |
| Mecanismos posibles |
|
| Posibilidad de ser evitado | Proveer datos sobre la posibilidad de prevenir el efecto adverso |
| Fuente de la evidencia | Ensayos
clínicos posautorización, estudios de seguridad, estudios
farmacoepidemiológicos, IPAS, otras notificaciones de seguridad |
1.2.4. Interacciones con otros medicamentos, alimentos, fitoterapéuticos
Para cada interacción importante proveer la siguiente información
| Sustancia con la que existe interacción |
|
| Efecto de la interacción |
|
| Fuente de la evidencia |
|
| Posible mecanismo |
|
| Riesgo potencial |
|
| Discusión |
|
1.2.5. Epidemiología de la(s) indicación(es)
1.2.5.1. Para cada indicación de uso discutir la incidencia,
prevalencia, mortalidad y perfil demográfico de la población objetivo
| Indicación/Población objetivo |
|
| Incidencia de la población objetivo |
|
| Prevalencia de la población objetivo |
|
| Mortalidad de la población objetivo |
|
| Perfil demográfico de la población objetivo | Proveer distribución por edad y sexo |
1.2.5.2. Para cada indicación de uso discutir las co-morbilidades importantes en la población objetivo
| Indicación/Población objetivo | Nombrar
la co-morbilidad en la población objetivo. Para cada co-morbilidad
importante, presentar incidencia, prevalencia y mortalidad en la
población objetivo y los principales medicamentos indicados en ese caso. |
1.2.5.3. Para cada riesgo identificado o potencial (ej.: falla
hepática) presentar la epidemiología de la condición en la población
objetivo cuando no fue expuesta al medicamento
| Riesgo potencial o identificado |
|
| Incidencia de la condición |
|
| Prevalencia de la condición |
|
| Mortalidad de la condición |
|
1.2.6. Efectos adversos de clase farmacológica
Identificar los riesgos que se cree que son comunes a la clase
farmacológica. Si un riesgo que es común a la clase farmacológica no se
piensa que es un problema de seguridad con el medicamento esto debe ser
justificado.
1.2.7. Otros requerimientos
Sobredosis potencial
Potencial de transmisión de agentes infecciosos
Potencial de uso ilegal
Potencial uso fuera de prospecto
Potencial uso fuera de prospecto en poblaciones pediátricas
Otros
1.3. Resumen
| Riesgos identificados importantes | Listado |
| Riesgos potenciales importantes | Listado |
| Información faltante importante | Listado |
2. Plan de farmacovigilancia
El plan de farmacovigilancia abarca acciones destinadas a identificar o
caracterizar los problemas de seguridad. No debe incluir acciones
destinadas a reducir o prevenir los riesgos.
2.1. Farmacovigilancia de rutina
Resumir brevemente el sistema de farmacovigilancia de rutina (ej.:
gestión de notificaciones espontáneas, presentación de IPAS, revisión
de bibliografía científica nacional e internacional).
2.2. Resumen de los problemas de seguridad y de las acciones de farmacovigilancia (rutina y adicionales)
Para cada problema de seguridad presentar un resumen de las acciones de
farmacovigilancia de rutina y adicionales. Las actividades adicionales
de farmacovigilancia pueden consistir, por ejemplo, en
farmacovigilancia activa o estimulada y realización de estudios
poscomercialización en Argentina (ejemplo: estudio de utilización de
medicamentos, centro centinela, estudios observacionales, entre otros).
Justificar cuando no se requieran actividades adicionales de farmacovigilancia.
| Problema de seguridad | Acciones de farmacovigilancia |
| Riesgos identificados importantes | Listado |
| Riesgos potenciales importantes | Listado |
| Información faltante importante | Listado |
2.3. Descripción del plan de farmacovigilancia para los problemas de seguridad
| Problema de seguridad | < > |
| Acción(es) propuesta(s) | <Si entre las acciones propuestas se encuentran estudios poscomercialización, completar el Anexo 2> |
| Objetivo de la(s) acción(es) propuesta(s) | < > |
| Justificación de la(s) acción(es) propuesta(s) | < > |
| Descripción
de nuevas medidas que puedan adaptarse en base a los resultados de la
acción propuesta y el criterio de decisión para iniciar dicha medida | < > |
| Métodos y períodos de tiempo para la evaluación y la presentación de informes a la Autoridad Regulatoria | < > |
3. Evaluación de la necesidad de las acciones de minimización de riesgo
Se debe evaluar la necesidad de las actividades de minimización de
riesgo para todos los riesgos listados en el punto 1.3, y justificar si
las acciones de minimización de riesgo de rutina (es decir la
información de seguridad contenida en rótulos y prospectos) son
suficientes o si son necesarias acciones adicionales de minimización de
riesgo (ejemplo: material educativo o programas de formación para
médicos, farmacéuticos y pacientes, programa de acceso restringido al
medicamento). Si se requieren actividades de minimización de riesgo
adicionales presentar un plan de minimización de riesgo.
Justificar cuando no se requieran actividades adicionales de minimización de riesgo.
3.1. Para cada problema de seguridad del punto 1.3 presentar un resumen
de las actividades de minimización de riesgo planificadas
| Problema de seguridad | ¿Las acciones de minimización de riesgo de rutina son suficientes | Justificación |
| Riesgos identificados importantes |
|
|
| <Listado de los riesgos> | Sí/No | < > |
| Riesgos potenciales importantes |
|
|
| <Listado de los riesgos> | Sí/No | < > |
| Información faltante importante |
|
|
| <Listado de los riesgos> | Sí/No | < > |
3.2. Errores de medicación potenciales
El TARC debe considerar sistemáticamente la posibilidad de que se
cometan errores de medicación teniendo en cuenta las fuentes de error
de medicación más comunes. Esta evaluación debe realizarse desde la
fase de desarrollo. El TARC debe discutir los posibles errores de
medicación, su posible causa y las acciones a tomar para evitar los
mismos. Entre los ítems a considerar se encuentran, por ejemplo, el
nombre comercial (contemplar la posibilidad de confusión con otras
especialidades medicinales); arte de los rótulos (diferenciación visual
de concentraciones diferentes de una misma especialidad medicinal);
tamaño, forma y color de la forma farmacéutica (confusión con otras
especialidades medicinales); instrucciones de uso (por dificultad en la
reconstitución, en la vía de administración parenteral, en el cálculo
de dosis). Si después de la comercialización, se evidencia que las
reacciones adversas se producen como consecuencia de los errores de
medicación, debe discutirse el tema en la actualización del PGR.
4. Plan de minimización de riesgo
Para cada riesgo identificado o potencial importante para el que se
necesiten acciones de minimización de riesgo adicionales, presentar:
| Problema de seguridad | <> |
| Actividades de minimización de riesgo de rutina (ej.: información de seguridad en prospecto, rótulo) | Breve descripción del texto que se incluirá en el prospecto sobre dicho riesgo. |
| 1-
Acción de minimización de riesgo adicional (ej.: material educativo,
programas de formación para médicos, farmacéuticos y pacientes) | Objetivo y justificación |
| Acciones propuestas |
| Métodos y criterios para evaluar la efectividad de las acciones propuestas |
| Período de revisión e informe a la Autoridad Regulatoria |
| 2- Acción de minimización de riesgo adicional (ej.: programa de acceso restringido) | Objetivo y justificación |
| Acciones propuestas |
| Criterios para evaluar la efectividad de las acciones propuestas |
| Período de revisión e informe a la Autoridad Regulatoria |
5. Resumen de las actividades del PGR
| Problema de seguridad | Actividades de farmacovigilancia propuestas (rutina y adicionales) | Actividades de minimización de riesgo propuestas (rutina y adicionales) |
| Problema de seguridad 1 | Ej.:
- Farmacovigilancia de rutina
- Estudio de utilización del medicamento para investigar... | Ej.:
- El problema de seguridad 1 aparece en el ítem de Contraindicaciones en el prospecto
- En el ítem precauciones dice...
- Material educativo
- Programa de acceso restringido (completar Anexo 3) |
| Problema de seguridad 2 |
|
|
6. Persona de contacto para este PGR
| Nombre |
|
| Cargo/función que ocupa en la empresa |
|
| Teléfono |
|
| Correo electrónico |
|
7. Presentación de documentos actualizados
Especificar el plazo de tiempo en que se presentará la actualización del documento con los resultados del PGR aplicado.
Anexo 1 Resumen de los protocolos de estudio de fase IV que se realizan en Argentina
| Título del estudio | < > |
| Fecha de inicio y finalización del estudio | < > |
| Estado del protocolo | < > |
| Objetivo del estudio | < > |
| Diseño y metodología del estudio | < > |
Anexo 2 Detalles del plan de minimización riesgo
En el caso de presentar un programa de acceso restringido describir el
circuito, los requisitos de cada paso (ej.: presentación de
documentación) y responsabilidades de los actores en la cadena de
comercialización (ej.: presentación por parte del TAR a la Autoridad
Regulatoria de un informe periódico con datos sobre la venta y el uso
de la especialidad medicinal).
Formulario 10. Versión 1
ANEXO III
GLOSARIO
Alerta. Señal que se considera lo suficientemente importante como para ser comunicada con cierta rapidez.
Base de datos de Farmacovigilancia. Sistema informático que permite el
registro de notificaciones de sospechas de reacciones adversas una vez
evaluadas y codificadas. Es el instrumento fundamental para la
generación de señales y, posteriormente, posibles alertas.
Beneficio. Habitualmente se expresa como el efecto terapéutico
demostrado que tiene un producto, aunque también debe incluir la
valoración subjetiva del paciente acerca de estos efectos.
Causa alternativa. En la evaluación de la causalidad, la existencia de
una explicación, una patología de base u otra medicación tomada
simultáneamente, más verosímil que la relación causal con el
medicamento evaluado.
Causalidad (ver también Imputabilidad). Resultado del análisis de la
imputabilidad y de la evaluación individual de la relación entre la
administración de un medicamento y la aparición de una reacción
adversa. Lleva a determinar una categoría de causalidad.
Categorías de causalidad. Las categorías descriptas por el Centro de Monitoreo de Uppsala son las siguientes:
a) Definida. Un acontecimiento clínico, incluyendo alteraciones en las
pruebas de laboratorio, que se manifiesta con una secuencia temporal
plausible en relación con la administración del medicamento, y que no
puede ser explicado por la enfermedad concurrente, ni por otros
medicamentos o sustancias. La respuesta a la supresión del medicamento
(retirada) debe ser plausible clínicamente. El acontecimiento debe ser
definitivo desde un punto de vista farmacológico o fenomenológico,
utilizando, si es necesario, un procedimiento de re-exposición
concluyente.
b) Probable. Acontecimiento clínico, incluyendo alteraciones en las
pruebas de laboratorio, que se manifiesta con una secuencia temporal
razonable en relación con la administración del medicamento, que es
improbable que se atribuya a la enfermedad concurrente, ni a otros
medicamentos o sustancias, y que al retirar el medicamento se presenta
una respuesta clínicamente razonable. No se requiere tener información
sobre re-exposición para asignar esta definición.
c) Posible. Acontecimiento clínico, incluyendo alteraciones en las
pruebas de laboratorio, que se manifiesta con una secuencia temporal
razonable en relación con la administración del medicamento, pero que
puede ser explicado también por la enfermedad concurrente, o por otros
medicamentos o sustancias. La información respecto a la retirada del
medicamento puede faltar o no estar clara.
d) No relacionada. Acontecimiento clínico, incluyendo alteraciones en
las pruebas de laboratorio, que se manifiesta con una secuencia
temporal improbable en relación con la administración del medicamento,
y que puede ser explicado de forma más plausible por la enfermedad
concurrente, o por otros medicamentos o sustancias.
e) Condicional. La secuencia temporal es razonable y la reacción no se
explicaría por el estado clínico del paciente, pero el cuadro
presentado no es conocido como efecto indeseable del medicamento
implicado. También es un acontecimiento clínico, incluyendo
alteraciones en las pruebas de laboratorio, notificado como una
reacción adversa, de la que es imprescindible obtener más datos para
poder hacer una evaluación apropiada, o los datos adicionales están
bajo examen.
f) Desestimada. Una notificación que sugiere una reacción adversa, pero
que no puede ser juzgada debido a que la información es insuficiente o
contradictoria, y que no puede ser verificada o completada en sus datos.
Clasificación Anatómica, Terapéutica y Química (ATC). Sistema de
codificación de los principios activos, según el órgano o sistema en el
que actúan y sus propiedades terapéuticas, farmacológicas y químicas.
Las drogas se clasifican en grupos en cinco niveles diferentes. El
primer nivel incluye 14 grandes grupos de sistemas/órganos. Cada uno de
estos grupos (primer nivel) está subdividido hasta cuatro niveles más;
el segundo y el tercer nivel forman subgrupos farmacológicos y
terapéuticos; el cuarto determina subgrupos
terapéutico/farmacológico/químicos, y el quinto designa cada fármaco.
Desvío de calidad. Fallas en las especificaciones que deben cumplir las
especialidades medicinales originadas durante su manufactura, siendo el
laboratorio productor responsable de ellas.
Efecto adverso. Ver Reacción adversa al medicamento.
Efecto colateral. (ver Reacción adversa al medicamento). Cualquier
efecto no intencionado de un producto farmacéutico que se produzca con
dosis normales utilizadas en el hombre, y que esté relacionado con las
propiedades farmacológicas del medicamento. Los elementos esenciales en
esta definición son la naturaleza farmacológica del efecto, que el
fenómeno no es intencionado y que no existe sobredosis evidente.
Efector periférico. Es la unidad funcional vinculada al sistema
sanitario, responsable de la realización de los programas oficiales de
Farmacovigilancia en un área determinada: programación, coordinación,
recolección, evaluación, codificación, formación, e información sobre
reacciones adversas a los medicamentos y desvíos de calidad.
Efectos adversos tipo A. Son aquellos debidos a los efectos
farmacológicos (aumentados). Tienden a ser bastante frecuentes,
dosis-dependientes y, a menudo, pueden ser evitados usando dosis más
apropiadas para el paciente individual. Estos efectos pueden
normalmente ser reproducidos y estudiados experimentalmente y,
frecuentemente, están ya identificados antes de su comercialización.
Efectos adversos tipo B. Característicamente suceden sólo en una
minoría de pacientes y muestran una mínima o ninguna relación con la
dosis. Normalmente son poco frecuentes e impredecibles, y pueden ser
graves y difíciles de estudiar. Pueden ser inmunológicos o manifestarse
solamente en algunos pacientes con factores predisponentes, a menudo
desconocidos. Las reacciones de tipo inmunológico pueden variar desde
erupciones (rash), anafilaxia, vasculitis, lesión orgánica
inflamatoria, hasta síndromes autoinmunes muy específicos. También se
presentan efectos de Tipo B no inmunológicos en una minoría de
pacientes predispuestos, intolerantes, por ejemplo, debido a un defecto
congénito del metabolismo o a una deficiencia adquirida de una enzima
determinada, con el resultado de una vía metabólica alterada o a una
acumulación de un metabolito tóxico.
Efectos adversos tipo C. Se refiere a situaciones en las que la
utilización del medicamento, a menudo por razones desconocidas, aumenta
la frecuencia de una enfermedad “espontánea”. Los efectos Tipo C pueden
ser graves y frecuentes (incluyen tumores malignos) y pueden ocasionar
efectos acusados en la salud pública. Pueden ser coincidentes, y a
menudo, estar relacionados, con efectos prolongados; frecuentemente no
hay secuencia temporal sugerente y puede ser difícil de probar la
asociación con el medicamento.
Efectos adversos tipo D. Incluyen la carcinogénesis y la teratogénesis.
Efecto adverso serio. Es aquel que provoca la muerte o amenaza de vida,
requiere o prolonga la hospitalización, produce una anomalía congénita
o deja una secuela permanente.
Efecto secundario. Efecto que no surge como consecuencia de la acción
farmacológica primaria de un medicamento, sino que constituye una
consecuencia eventual de esta acción, por ejemplo, la diarrea asociada
con la alteración del equilibrio de la flora bacteriana normal que es
producto de un tratamiento antibiótico. En sentido estricto, este
término no debe emplearse como sinónimo de efecto colateral.
ESAVI: Evento adverso supuestamente atribuible a la vacunación o inmunización.
Según el componente que las produce, pueden ser:
Reacciones al antígeno inmunizante: la fiebre y el exantema después de
la administración de la vacuna antisarampionosa y el dolor al tacto, el
enrojecimiento y la tumefacción después de inyectar la vacuna contra la
fiebre tifoidea son ejemplos de reacciones adversas leves posteriores a
la inmunización. La linfadenitis causada por algunas cepas de BCG y la
aplicación de dosis de refuerzo de toxoide diftérico y tetánico en
personas con títulos altos. Dentro de las reacciones graves (escasas)
se encuentra la parálisis después de aplicar la vacuna
antipoliomielítica.
Reacciones a otros componentes de la vacuna: tales como antibióticos, agentes conservadores y adyuvantes.
Errores programáticos: se deberán a cualquier error en la conservación,
almacenaje, transportación y administración de la vacuna.
Se deben identificar las causas de ocurrencia de errores programáticos, entre ellas se cuentan:
a) Vacunas aplicadas en sitios incorrectos.
b) Uso de agujas y jeringas no esterilizadas.
c) Manipulación incorrecta de agujas.
d) Vacunas reconstituidas con diluyentes no apropiados.
e) Incremento de la dosis de vacunas.
f) Sustitución de vacunas por otros productos.
g) Vacunas y diluyentes contaminados.
h) Incorrecta conservación de la vacuna.
Evento adverso. Cualquier suceso médico desafortunado que puede
presentarse durante el tratamiento con un medicamento pero que no
necesariamente tiene una relación causal con dicho tratamiento. En este
caso ocurre la coincidencia en el tiempo sin ninguna sospecha de una
relación causal.
Excipiente. Sustancia desprovista de actividad farmacológica previsible
que se añade a un medicamento con el fin de darle una forma,
consistencia, olor, sabor o cualquier otra característica que lo haga
adecuado para su administración. En ocasiones los excipientes son causa
de efectos indeseados, sobre todo de tipo alérgico.
Falta de eficacia (fallo terapéutico, inefectividad terapéutica). Fallo
inesperado de un medicamento en producir el efecto previsto como lo
determinó previamente una investigación científica.
Farmacoepidemiología. Estudio del uso y los efectos de los medicamentos
en grandes poblaciones. Epidemiología del medicamento. Incluye estudios
de utilización de medicamentos, ensayos clínicos y farmacovigilancia.
Farmacovigilancia activa o estimulada. Son las actividades orientadas a
alentar a los profesionales de la salud a notificar reacciones
adversas. Estas acciones pueden estar incluidas dentro del PGR como
actividad de Farmacovigilancia adicional.
Farmacovigilancia intensiva. Es el monitoreo sistemático de la
aparición de eventos adversos de un principio activo durante toda la
etapa de prescripción (Disposición ANMAT N° 2552/95). Método de la
Farmacovigilancia que consiste en obtener información de sospechas de
reacciones adversas a medicamentos de manera sistemática, de calidad y
completa, caracterizada por su elevada sensibilidad y fiabilidad;
especialmente cuando se hace necesario determinar la frecuencia de las
reacciones adversas, identificar factores predisponentes, patrones de
uso de medicamentos, entre otros.
Hipersensibilidad. (ver Reacción alérgica al medicamento).
Hoja amarilla. Es el formulario donde se recopilan sospechas de
reacciones adversas, editado en color amarillo y distribuido por el
Programa Nacional de Farmacovigilancia a los profesionales sanitarios
que les permite la notificación. Recoge información relativa al
paciente (identificación, edad, sexo, peso), al medicamento sospechoso
(nombre, dosis, frecuencia, fecha de inicio y final, indicación
terapéutica), a la reacción adversa (descripción, fecha de comienzo y
final, desenlace, efecto de la reexposición si ha existido, etc.) y al
profesional notificador (nombre, dirección, teléfono, profesión, nivel
asistencial, etc.).
Hoja de notificación. Hoja amarilla, formulario CIOMS/Medwatch.
Imputabilidad. (Causalidad). Es el análisis, caso por caso, de la
relación de causalidad entre la administración de un medicamento y la
aparición de una reacción adversa. Se trata de un análisis individual
para una notificación dada, que no pretende estudiar el potencial de
riesgo del medicamento de forma global o la importancia del riesgo
inducido por el medicamento en la población. Los métodos de
imputabilidad sirven para armonizar y estandarizar el proceso de
imputación, y para permitir la reproducibilidad de un evaluador a otro.
Información faltante. Es información de seguridad del medicamento que
no está disponible en el momento de la presentación del medicamento y
representa una limitación de la información de seguridad.
Informe Periódico de Actualización de Seguridad (IPAS). Los informes
periódicos de actualización de seguridad son documentos que se crearon
con el fin de mantener información actualizada a nivel mundial de los
reportes y datos de seguridad de un producto comercializado. Reúnen la
experiencia acumulada a través de períodos de tiempo establecidos de
los eventos adversos reportados mundialmente y por ende permiten a las
autoridades sanitarias o a las compañías productoras prever las
necesidades de realizar nuevos estudios, hacer cambios en la
información básica de prescripción o tomar decisiones frente al mercado.
Medicamento de reciente comercialización. Todo medicamento que se
encuentre en sus primeros cinco años de comercialización (no
necesariamente coincidente con el plazo de su aprobación).
Notificación. (ver también Hoja amarilla). La comunicación de una
sospecha de reacción adversa a un medicamento a un centro de
Farmacovigilancia. Usualmente estas notificaciones se realizan mediante
los formularios de recopilación de una reacción adversa, procurando los
medios necesarios en cada caso para mantener la confidencialidad de los
datos.
Notificador. Toda persona que haya sospechado de una probable reacción
adversa a un medicamento y que lo haya comunicado a un Centro de
Farmacovigilancia.
Plan de Gestión de Riesgo. Es un conjunto de actividades e
intervenciones en Farmacovigilancia diseñadas para identificar,
caracterizar, prevenir o minimizar riesgos relacionados a productos
medicinales, y la evaluación de la efectividad de esas intervenciones.
PSUR. Periodic Safety Update Report (Ver IPAS).
Reacción adversa a medicamentos (RAM). Según la OMS, “reacción nociva y
no deseada que se presenta tras la administración de un medicamento, a
dosis utilizadas habitualmente en la especie humana, para prevenir,
diagnosticar o tratar una enfermedad, o para modificar cualquier
función biológica”. Nótese que esta definición implica una relación de
causalidad entre la administración del medicamento y la aparición de la
reacción. En la actualidad se prefiere “efecto no deseado atribuible a
la administración de...” y reservar la definición original de la OMS
para el concepto de acontecimiento adverso, el cual no implica
necesariamente el establecimiento de una relación de causa a efecto.
Nótese además que esta definición excluye las intoxicaciones o
sobredosis. Respuesta a un medicamento que es nociva y no intencionada,
y que se produce con las dosis utilizadas normalmente en el hombre. En
esta descripción es importante ver que se involucra la respuesta del
paciente, que los factores individuales pueden tener un papel
importante y que el fenómeno es nocivo (una respuesta terapéutica
inesperada, por ejemplo, puede ser un efecto colateral pero no ser una
reacción adversa).
Reacción adversa inesperada. Reacción que no ha sido descrita en el
rótulo y prospecto del producto o que no ha sido reportada a la
autoridad sanitaria por el laboratorio que obtuvo el registro del
producto al momento de solicitarlo (ver también Reacción adversa al
medicamento). Reacción adversa, cuya naturaleza o intensidad no es
consistente con la información local o la autorización de
comercialización, o bien no es esperable por las características
farmacológicas del medicamento. El elemento predominante en este caso
es que el fenómeno sea desconocido.
Reacción adversa seria. Ver Efecto adverso serio.
Reacción alérgica al medicamento. Reacción adversa al medicamento que
se caracteriza por ser dosis-independiente y que es mediada por el
sistema inmunológico.
Las reacciones alérgicas se han clasificado en cuatro tipos clínicos principales:
Tipo 1: conocido como reacción anafilactoide inmediata o de
hipersensibilidad inmediata, está mediada por la interacción del
alergeno (medicamento) y los anticuerpos de tipo IgE. Las reacciones
producidas por administración de la penicilina constituyen un ejemplo
de este tipo.
Tipo 2: o citotóxica, consisten en reacciones de fijación del
complemento entre el antígeno y un anticuerpo presente en la superficie
de algunas células. Estas reacciones incluyen las anemias hemolíticas
provocadas por medicamentos, las agranulocitosis y otras.
Tipo 3: reacción mediada por un complejo inmune que se deposita en las células del tejido u órgano blanco.
Tipo 4: resulta de la interacción directa entre el alergeno
(medicamento) y los linfocitos sensibilizados. También se conoce como
reacción alérgica retardada e incluye la dermatitis por contacto.
Reexposición. En la evaluación de la relación de causalidad, cuando la
reacción o acontecimiento aparecen de nuevo tras la administración del
medicamento sospechoso.
Riesgo. Es la probabilidad de ocasionar un perjuicio, que normalmente
expresa la probabilidad de un suceso como un porcentaje o una razón.
Riesgo identificado. Es un evento desfavorable para el que hay una
adecuada evidencia de asociación con el medicamento de interés. Son
ejemplos de riesgos identificados:
• Una reacción adversa adecuadamente demostrada en los estudios preclínicos y confirmada con información clínica.
• Una reacción adversa observada en ensayos clínicos o estudios
epidemiológicos en los que la magnitud de la diferencia con el grupo
comparador (placebo o sustancia activa, o grupo no expuesto), sobre un
parámetro de interés sugiere una relación causal.
• Una reacción adversa sugerida por un número de notificaciones
espontáneas bien documentadas en las que la causalidad está fuertemente
apoyada por una relación temporal y plausabilidad biológica, como las
reacciones anafilácticas en el sitio de aplicación.
Riesgo potencial. Es un evento desfavorable para el que existen algunas
bases de sospecha de asociación con el medicamento de interés pero la
asociación no ha sido confirmada. Los ejemplos de riesgo potencial son:
• Temas de seguridad preclínica que no han sido observados en los ensayos clínicos.
• Una reacción adversa observada en ensayos clínicos o estudios
epidemiológicos en los que la magnitud de la diferencia con el grupo
comparador (placebo o sustancia activa, o grupo no expuesto), sobre un
parámetro de interés sobre el que existe sospecha, no es
suficientemente grande como para sugerir una relación causal.
• Una señal que se origina en el sistema de reportes espontáneos.
• Un evento que se conoce porque se asocia con otros productos de la
misma clase o para el cual se espera que ocurra por las propiedades de
la especialidad medicinal.
Secuencia temporal. En la evaluación de la relación de causalidad,
valora el tiempo transcurrido entre el inicio del tratamiento y la
aparición de las primeras manifestaciones de la reacción.
Seguridad. Característica de un medicamento que puede usarse con una
probabilidad muy pequeña de causar efectos tóxicos injustificables. La
seguridad de un medicamento es por lo tanto una característica relativa
y en farmacología clínica su medición es problemática debido a la falta
de definiciones operativas y por razones éticas y legales.
Señal. Información comunicada de una posible relación causal entre un
acontecimiento adverso y un medicamento, cuando previamente esta
relación era desconocida o estaba documentada de forma incompleta. A su
vez, las señales pueden generar, posteriormente, alertas, dependiendo
de la gravedad del acontecimiento y de la calidad de la información.
Sistema de notificación espontánea. Método de Farmacovigilancia, basado
en la comunicación, recogida y evaluación de notificaciones realizadas
por un profesional sanitario, paciente o familiar, de sospechas de
reacciones adversas a medicamentos, dependencia de medicamentos, abuso
y mal uso de medicamentos.
Teratogenicidad. Capacidad del medicamento de causar daño en el embrión
o feto y, en un sentido estricto, malformaciones estructurales durante
cualquiera de las etapas de desarrollo.
Toxicidad. Grado en que una sustancia es nociva. Fenómenos nocivos
debidos a una sustancia o medicamento y observados después de su
administración.
Uppsala Monitoring Centre (UMC). Centro Internacional de Monitoreo de Medicamentos de Uppsala dependiente de la OMS.
WHO-ART (WHO Adverse Reaction Terminology). Diccionario de terminología
que contiene un sistema de codificación de reacciones adversas de
medicamentos.