MINISTERIO DE RELACIONES EXTERIORES Y CULTO
MERCOSUR/GMC/RES. Nº 01/15
REQUISITOS DE BUENAS PRÁCTICAS PARA EL FUNCIONAMIENTO DE LOS SERVICIOS DE SALUD
VISTO: El Tratado de Asunción y el Protocolo de Ouro Preto
CONSIDERANDO:
La necesidad de contar con Requisitos de Buenas Prácticas para el Funcionamiento de los servicios de salud.
GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar los Requisitos de Buenas Prácticas para el
Funcionamiento de los servicios de salud, que constan como Anexo y
forman parte de la presente Resolución.
Art. 2 - Los Requisitos de Buenas Prácticas establecidos en la presente
Resolución deben ser incluidos en las normas de organización y
funcionamiento de los servicios de salud de cada Estado Parte, pudiendo
agregarse otros requisitos a la normativa nacional o local de acuerdo
con la necesidad de cada Estado Parte.
Art. 3 - Los Estados Partes indicarán, en el ámbito del SGT N° 11, los
organismos nacionales competentes para la implementación de la presente
Resolución.
Art. 4 - Esta Resolución debe ser incorporada al ordenamiento jurídico de los Estados Partes, antes del 1/XII/2015.
XCVIII GMC - Brasilia, 29/V/15.
ANEXO
REQUISITOS DE BUENAS PRÁCTICAS PARA EL FUNCIONAMIENTO DE LOS SERVICIOS DE SALUD
1. OBJETIVOS
Establecer los Requisitos de Buenas Prácticas para el Funcionamiento de
los servicios de salud, entendiendo como tales los establecimientos
destinados al desarrollo de acciones relacionadas a la promoción,
protección, mantenimiento y recuperación de la salud, cualquiera sea el
nivel de complejidad, ya sea con régimen de internación o no,
incluyendo la atención realizada en consultorio o domicilio; basado en
la mejora de la calidad, en la humanización de la atención y su gestión.
Promover la reducción y control de riesgos a los usuarios, trabajadores de la salud y al medio ambiente.
2. GESTIÓN DE LA CALIDAD
2.1 Los servicios de salud deben desarrollar acciones para establecer
una política de calidad que involucre: estructura, proceso y resultado
en la gestión de los mismos.
2.1.1 El servicio de salud deberá utilizar la Garantía de la Calidad
como herramienta de gestión, entendiendo a la misma como la totalidad
de las acciones sistemáticas necesarias para garantizar que los
servicios prestados estén dentro de los estándares de calidad exigidos
para los fines que se proponen.
2.2 Las Buenas Prácticas de Funcionamiento son componentes de la
Garantía de la Calidad que aseguran que los servicios sean ofrecidos
con los estándares de calidad adecuados.
2.2.1 Las Buenas Prácticas de Funcionamiento están orientadas
principalmente a la reducción de riesgos inherentes a la prestación de
los servicios de salud.
2.2.2 Los conceptos de Garantía de la Calidad y Buenas Prácticas de
Funcionamiento aquí descriptos están relacionados entre sí con el fin
de enfatizar sus relaciones y su importancia para el buen
funcionamiento de los servicios de salud.
2.3 Las Buenas Prácticas de Funcionamiento determinan que:
2.3.1 El servicio de salud debe ser capaz de ofrecer servicios dentro
de los estándares de calidad requeridos, cumpliendo con los requisitos
de la normativa vigente en cada Estado Parte.
2.3.2 El servicio de salud debe proporcionar todos los recursos necesarios, incluyendo:
a) Personal calificado, capacitado e identificado;
b) Ambientes identificados;
c) Equipo, material y apoyo logístico; y
d) Los procedimientos e instrucciones aprobadas y vigentes.
2.3.3 Las quejas sobre los servicios prestados deben ser examinadas y
registradas y las causas de los desvíos de calidad deben ser
investigadas y documentadas debiéndose tomar medidas respecto a los
servicios con desvío de la calidad, adoptando las medidas para prevenir
recurrencias.
3. LA SEGURIDAD DEL PACIENTE
3.1 El servicio de salud debe establecer estrategias y acciones direccionadas para la seguridad del paciente, tales como:
3.1.1 Mecanismos de identificación del paciente;
3.1.2 Orientaciones para la higienización de las manos;
3.1.3 Acciones para prevenir y controlar los efectos adversos relacionados con la atención a la salud;
3.1.4 Mecanismos para garantizar la seguridad quirúrgica;
3.1.5 Orientaciones para la administración segura de medicamentos, sangre, productos sanguíneos y hemocomponentes;
3.1.6 Mecanismos para la prevención de caída de los pacientes;
3.1.7 Mecanismos para la prevención de úlceras por presión;
3.1.8 Orientaciones para fomentar la participación del paciente en la atención prestada.
4. CONDICIONES DE LA ORGANIZACIÓN
4.1 El servicio de salud debe tener estatuto interno o documento
equivalente actualizado, teniendo en cuenta la definición y descripción
de todas las actividades técnicas, administrativas y asistenciales, las
responsabilidades y atribuciones.
4.2 Los servicios de salud deben poseer una habilitación o una licencia
de funcionamiento, actualizada periódicamente, emitida por la autoridad
sanitaria competente.
4.3 Los servicios y actividades tercerizados por los establecimientos de salud deben tener contratos de prestación de servicios.
4.3.1 Los servicios y actividades tercerizados deben estar autorizados
por la autoridad sanitaria competente, cuando así lo establezcan las
normativas vigentes en cada Estado Parte.
4.3.2 La habilitación de los servicios y actividades tercerizados
deberá informar acerca de su capacidad para prestar este servicio,
conforme las normativas vigentes en cada Estado Parte.
4.4 El alcance de los estándares sanitarios establecidos por esta
resolución no exime al servicio de salud en el cumplimiento de las
demás normativas vigentes en cada Estado Parte.
4.5 El servicio de salud debe tener un responsable técnico y un
reemplazante, legalmente autorizados por la autoridad competente.
4.5.1 La autoridad competente deberá ser notificada siempre que haya cambios del responsable técnico o su reemplazante.
4.6 La dirección y el responsable técnico del servicio de salud tienen
la responsabilidad de planificar, ejecutar y asegurar la calidad de los
procesos.
4.7 Las unidades funcionales de los servicios de salud deben tener un
profesional a cargo, conforme esté definido en las normativas
específicas vigentes en cada Estado Parte.
4.8 El servicio de salud debe poseer un profesional legalmente
habilitado que responda a las cuestiones operacionales durante el
período de funcionamiento.
4.9 El servicio de salud debe proporcionar la infraestructura física,
recursos humanos, equipos, suministros y materiales necesarios a la
operacionalización del servicio de acuerdo a la capacidad, el tipo de
asistencia ofrecida y a las normativas vigentes en cada Estado Parte.
4.10 El servicio de salud debe tener mecanismos para garantizar la
continuidad de la atención al paciente cuando haya la necesidad de
traslado o la realización de exámenes que no estén disponibles en el
servicio.
4.10.1 Todo paciente trasladado debe ser acompañado por lo menos con un
resumen completo de su historia clínica, legible, con identificación y
firma del profesional asistente, que debe formar parte de la historia
clínica en el destino, quedando una copia en la historia clínica de
origen.
4.11 El servicio de salud debe poseer mecanismos para garantizar el
funcionamiento de las Comisiones, Comités y Programas establecidos en
la normativa vigente en cada Estado Parte.
4.12 El servicio de salud debe tener mecanismos para garantizar el
control del acceso de los trabajadores, pacientes, acompañantes y
visitantes.
4.13 El servicio de salud debe tener mecanismos para garantizar la
identificación de los trabajadores, pacientes, acompañantes y
visitantes.
4.14 El servicio de salud debe mantener disponible, según el tipo de actividad, la documentación y registros relativos a:
4.14.1 Plan de desarrollo arquitectónico aprobado por la autoridad competente.
4.14.2 Control de la salud de los trabajadores;
4.14.3 Educación permanente de los profesionales;
4.14.4 Comisiones, comités y programas existentes;
4.14.5 Contratos de servicios tercerizados;
4.14.6 Control de la calidad del agua;
4.14.7 Mantenimiento preventivo y correctivo de la construcción e instalaciones;
4.14.8 Control de vectores y plagas urbanas;
4.14.9 Mantenimiento correctivo y preventivo de los equipamientos e instrumentos;
4.14.10 Plan de Gestión de Residuos de los servicios de salud;
4.14.11 Nacimientos;
4.14.12 Muertes;
4.14.13 Admisiones y altas;
4.14.14 Eventos adversos y quejas técnicas relacionados con productos o servicios;
4.14.15 Monitoreo e relatos referentes al control de infecciones;
4.14.16 Enfermedades de Declaración Obligatoria;
4.14.17 Indicadores previstos en las normativas existentes;
4.14.18 Normas, rutinas y procedimientos utilizados en el servicio;
4.14.19 Otros documentos exigidos por la normativa vigente en cada Estado Parte.
5. HISTORIA CLÍNICA DEL PACIENTE
5.1 La responsabilidad por el registro en la historia clínica es de
todos los profesionales de la salud que prestan el cuidado al paciente.
5.2 La custodia de la historia clínica es responsabilidad del servicio
de salud, que debe cumplir con las normativas vigentes en cada Estado
Parte.
5.2.1 El servicio de salud debe garantizar la custodia de la historia
clínica fundamentalmente respecto a la confidencialidad e integridad de
la misma.
5.2.2 El servicio de salud debe mantener la historia clínica en ámbito
seguro, en buenas condiciones de conservación y organización, que
permita el acceso cuando sea necesario.
5.3 El servicio de salud debe garantizar que la historia clínica
contenga los registros relativos a los datos identificatorios y a todas
las prestaciones realizadas al paciente.
5.4 El servicio de salud debe garantizar que la historia clínica deberá
ser escrita de forma legible por todos los profesionales directamente
involucrados en la atención al paciente, identificándose los mismos con
la firma y sello aclaratorio en sus intervenciones, en caso de historia
clínica en medio físico.
5.5 La historia clínica pertenece al paciente y deberá estar
permanentemente disponible para el mismo o sus representantes legales y
para la autoridad competente, cuando sea necesario.
6. GESTIÓN DE PERSONAL
6.1 Los requisitos relativos a los recursos humanos del servicio de
salud incluyen a los profesionales de todos los niveles de formación,
cualquiera sea la modalidad de relación laboral con el servicio.
6.2 El servicio de salud debe contar con equipo multidisciplinario acorde a la actividad que realizan y su perfil de demanda.
6.3 El servicio de salud debe mantener disponibles los registros de
capacitación de los profesionales compatibles con las funciones
ejercidas.
6.3.1 El servicio de salud debe contar con la documentación relativa a
la habilitación de los profesionales para su ejercicio cuando fuera
necesario.
6.4 El servicio de salud debe promover la capacitación de sus
profesionales al inicio de las actividades y de modo periódico, de
acuerdo con el perfil del servicio, cuando sea necesario.
6.4.1 Las capacitaciones deben ser registradas contemplando toda la información relativa a su contenido y modalidad.
6.5 La capacitación referida en el artículo anterior debe adaptarse a
la evolución del conocimiento y la identificación de nuevos riesgos.
7. GESTIÓN DE INFRAESTRUCTURA
7.1 El servicio de salud debe contar con un plan de desarrollo
arquitectónico actualizado, acorde a las actividades desarrolladas y
aprobado por los órganos competentes.
7.2 Las instalaciones de agua, alcantarillado, electricidad, gas, aire,
protección y extinción de incendios, comunicación y las demás
existentes, deben cumplir con los requisitos de los códigos de
construcción y reglamentos locales, así como las normas técnicas
aplicables a cada una de las instalaciones.
7.3 El servicio de salud debe mantener las instalaciones físicas de los
ambientes externos e internos en buenas condiciones de conservación,
seguridad, organización, comodidad y limpieza.
7.4 El servicio de salud debe ejecutar acciones de gestión de riesgos de accidentes inherentes a las actividades desarrolladas.
7.5 El servicio de salud debe poseer iluminación y ventilación en consonancia con el desarrollo de sus actividades.
7.6 El servicio de salud debe garantizar la calidad del agua necesaria para el funcionamiento de sus unidades.
7.6.1 El servicio de salud debe garantizar que la limpieza de los
depósitos de agua sea realizada conforme las normativas vigentes en
cada Estado Parte.
7.6.2 El servicio de salud debe mantener los registros de la capacidad y de la limpieza periódica de los depósitos de agua.
7.7 El servicio de salud debe garantizar la continuidad del suministro
de agua, incluso en el caso de interrupción del suministro por el
proveedor, en los lugares de los servicios donde el agua se considera
insumo crítico.
7.8 El servicio de salud debe garantizar la continuidad del suministro
de energía eléctrica en caso de interrupción del suministro por el
proveedor a través de sistemas de alimentación de emergencia, en los
lugares de los servicios donde la electricidad se considera insumo
crítico.
7.9 El servicio de salud debe tomar medidas para el mantenimiento
preventivo y correctivo de las instalaciones del edificio, por medio
propio o de terceros.
7.10 El servicio debe garantizar condiciones de accesibilidad en todos
sus sectores, incluyendo servicios sanitarios, para los discapacitados.
8. PROTECCIÓN DE LA SALUD DE LOS TRABAJADORES
8.1 El servicio de salud debe garantizar los mecanismos respecto a la
inmunización contra agentes biológicos a que los trabajadores puedan
estar expuestos.
8.2 El servicio de salud debe asegurar que los empleados sean evaluados
periódicamente en relación a su estado de salud, manteniendo registros
de esa evaluación.
8.3 El servicio de salud debe garantizar que los trabajadores con
problemas de salud solamente se reintegren a sus actividades habituales
después del alta médica.
8.4 El servicio de salud debe garantizar que sus trabajadores, con
exposición potencial a riesgos biológicos, físicos o químicos utilicen
indumentaria de trabajo, incluyendo calzado, que sean compatibles con
el riesgo y en condiciones de confort.
8.5 El servicio de salud debe garantizar los mecanismos para prevenir
el riesgo de accidentes de trabajo, incluyendo la provisión de Equipos
de Protección Individual (EPI) en número suficiente y compatible con
las actividades realizadas por los trabajadores.
8.5.1 Los trabajadores no deben dejar el lugar de trabajo con los equipos de protección individual.
8.6 El servicio de salud debe mantener los registros de comunicación de accidentes de trabajo.
8.7 El Servicio de Salud debe mantener disponible para todos los empleados, según sus funciones:
8.7.1 Normas y conductas de seguridad biológica, química, física, ocupacional y del ambiente;
8.7.2 Instrucciones para el uso de los Equipos de Protección Individual (EPI);
8.7.3 Procedimientos en caso de incendios y accidentes;
8.7.4 Orientación para el manejo y transporte de productos para la salud contaminados.
9. GESTIÓN DE TECNOLOGÍAS Y PROCESOS
9.1 El servicio de salud debe contar con normas, procedimientos y
rutinas técnicas escritas y actualizadas, de los procesos de trabajo en
un lugar de fácil acceso a todo el equipo.
9.2 El servicio de salud debe mantener los ambientes limpios, libres de
residuos y olores que sean incompatibles con la actividad y debe
cumplir con los criterios de criticidad para las áreas.
9.3 El servicio de salud debe garantizar la disponibilidad de los
equipos, materiales, insumos y medicamentos en acuerdo con la
complejidad, el tipo de servicio y la cantidad de usuarios atendidos en
el servicio.
9.4 El servicio de salud debe llevar a cabo la gestión de sus
tecnologías para satisfacer las necesidades del servicio, manteniendo
las condiciones de selección, adquisición, almacenamiento, instalación,
operación, distribución, eliminación y trazabilidad para esos fines.
9.5 El servicio de salud debe garantizar que los materiales y equipos
sean utilizados exclusivamente para los fines a que están destinados.
9.6 El servicio de salud debe garantizar que los colchones y almohadas
estén protegidos con material impermeable y lavable, y que no presenten
deterioros.
9.7 El servicio de salud debe garantizar la calidad de los
procedimientos de desinfección y esterilización de equipos y materiales.
9.8 El servicio de salud debe garantizar que todos los usuarios reciban soporte vital inmediato cuando sea necesario.
9.9 El servicio de salud debe proporcionar los insumos, productos y
equipos necesarios para las prácticas de higiene de manos de los
trabajadores, pacientes, acompañantes y visitantes.
9.10 El servicio de salud que brinde asistencia nutricional o comidas
debe garantizar la calidad nutricional y la seguridad de los alimentos.
9.11 El servicio de salud debe informar a los órganos competentes sobre
la sospecha de enfermedad de notificación obligatoria, según lo
establecido en las normativas vigentes en cada Estado Parte.
9.12 El servicio de salud deberá calcular y mantener los registros
relacionados a los indicadores establecidos en las normativas vigentes
en cada Estado Parte.
10. CONTROL INTEGRADO DE VECTORES Y PLAGAS URBANAS
10.1 El servicio de salud debe garantizar acciones eficaces y continuas
de control de vectores y plagas urbanas, de acuerdo con la normativa
vigente en los Estados Partes.
10.2 No se permite comer o almacenar alimentos en el lugar de trabajo
designado para la ejecución de los procedimientos de salud.
MERCOSUR/GMC/RES. Nº 02/15
REQUISITOS DE BUENAS PRÁCTICAS PARA
ORGANIZACIÓN Y FUNCIONAMIENTO DE SERVICIOS DE URGENCIA Y EMERGENCIA
(DEROGACIÓN DE LA RES. GMC N° 12/07)
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto y la Resolución N° 12/07 del Grupo Mercado Común.
CONSIDERANDO:
La necesidad de contar con Requisitos de Buenas Prácticas para
organización y funcionamiento de los servicios de urgencia y emergencia.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar los Requisitos de Buenas Prácticas para organización y
funcionamiento de servicios de urgencia y emergencia, que constan como
Anexo y forman parte de la presente Resolución.
Art. 2 - Los Requisitos de Buenas Prácticas establecidos en la presente
Resolución se aplican a la atención en servicios de urgencia y
emergencia y no se aplican a la atención móvil pre- hospitalaria.
Art. 3 - Los Requisitos de Buenas Prácticas establecidos en la presente
Resolución deben ser incluidos en las normas de organización y
funcionamiento de los servicios de urgencia y emergencia de cada Estado
Parte, pudiendo agregarse otros requisitos a la normativa nacional o
local de acuerdo con la necesidad de cada Estado Parte.
Art. 4 - Los Estados Partes indicarán, en el ámbito del SGT N° 11, los
organismos nacionales competentes para la implementación de la presente
Resolución.
Art. 5 - Derogar la Resolución GMC N° 12/07.
Art. 6 - Esta Resolución debe ser incorporada al ordenamiento jurídico de los Estados Partes, antes del 1/XII/2015.
XCVIII GMC - Brasilia, 29/V/15.
ANEXO
REQUISITOS DE BUENAS PRÁCTICAS PARA ORGANIZACIÓN Y FUNCIONAMIENTO DE SERVICIOS DE URGENCIA Y EMERGENCIA
1. OBJETIVO
Establecer los Requisitos de Buenas Prácticas para organización y funcionamiento de servicios de urgencia y emergencia.
2. DEFINICIONES
2.1 Emergencia: Verificación médica de condiciones de daño a la salud
que impliquen sufrimiento grave o riesgo inminente de muerte lo que
requiere tratamiento médico inmediato.
2.2 Urgencia: Ocurrencia de problemas de salud inesperados con o sin
riesgo potencial de muerte, en la que el individuo requiere atención
médica inmediata.
3. REQUISITOS
3.1 El servicio de urgencia y emergencia fijo puede funcionar como un
servicio de salud independiente o inserto en un establecimiento con
internación con mayor capacidad de resolución.
3.1.1. Los servicios de urgencia y emergencia deben estar organizados y
estructurados considerando las necesidades de la red de atención a la
salud existente.
3.2 Todo servicio de urgencia y emergencia, público o privado, debe
poseer o estar inserto en un servicio de salud que posea la
habilitación o licencia de funcionamiento, expedida por el órgano
sanitario competente, de acuerdo con la normativa de cada Estado Parte.
3.3 La construcción, reforma o adaptación a la estructura física del
servicio de urgencia y emergencia debe ser precedida del análisis y
aprobación del proyecto junto al órgano competente.
3.3.1. El órgano sanitario competente debe verificar la ejecución de las obras conforme a lo aprobado.
3.4 Es de responsabilidad de la administración del servicio de salud
prever y proveer los recursos humanos, equipamientos, materiales y
medicamentos necesarios para el funcionamiento de los servicios de
urgencia y emergencia.
3.5 La dirección del servicio de salud y el jefe del servicio de
urgencia y emergencia tienen la responsabilidad de planear, implementar
y garantizar la calidad de los procesos.
3.6 El servicio de urgencia y emergencia debe disponer de instrucciones
escritas y actualizadas de las rutinas técnicas implementadas.
3.7 Las rutinas técnicas deben ser elaboradas en conjunto con las áreas
involucradas en la asistencia al paciente, asegurando la asistencia
integral y la interdisciplinariedad.
3.8 El servicio de urgencia y emergencia debe:
3.8.1 poseer estructura organizacional documentada;
3.8.2 preservar la identidad y la privacidad del paciente, asegurando un ambiente de respeto y dignidad;
3.8.3 promover un ambiente acogedor;
3.8.4 ofrecer orientación al paciente y a los familiares en lenguaje
claro, sobre el estado de salud y la asistencia a ser prestada, desde
la admisión hasta el alta.
4. RECURSOS HUMANOS
4.1 Todo servicio de urgencia y emergencia debe disponer de los siguientes profesionales de la salud:
4.1.1 Responsable Técnico legalmente habilitado;
4.1.1.1 El responsable técnico puede asumir la responsabilidad por un (01) servicio de urgencia y emergencia;
4.1.1.2 En caso de ausencia del responsable técnico, el servicio debe contar con un profesional legalmente habilitado;
4.1.2 Todo servicio de urgencia y emergencia debe disponer de equipo médico en cantidad suficiente para la atención 24 horas;
4.1.2.1 El servicio de urgencia y emergencia de mayor complejidad debe
contar con profesionales especializados de acuerdo con el perfil de
atención, capacitados para atención de las urgencias y emergencias;
4.1.3 Enfermero exclusivo de la unidad, responsable para la coordinación de la asistencia de enfermería;
4.1.3.1 Equipo de enfermería en cantidad suficiente para la atención a
las 24 horas del día en todas las actividades correspondientes.
4.2 Todos los profesionales de los servicios de urgencia y emergencia
deben ser vacunados de acuerdo a la normativa nacional vigente.
4.3 El servicio de urgencia y emergencia debe promover entrenamiento y
educación permanente en conformidad a las actividades desarrolladas, a
todos los profesionales involucrados en la atención de pacientes,
manteniendo disponibles los registros de su realización y de la
participación de estos profesionales.
5. INFRAESTRUCTURA FÍSICA
5.1 El servicio de urgencia y emergencia debe disponer de
infraestructura física dimensionada de acuerdo a la demanda,
complejidad y perfil asistencial de la unidad, garantizando la
seguridad y continuidad de la asistencia al paciente;
5.1.1 El servicio de urgencia y emergencia debe garantizar, conforme al
perfil asistencial, el acceso independiente para pediatría.
5.2 El servicio de urgencia y emergencia debe poseer de acuerdo al perfil de atención, los siguientes ambientes:
5.2.1 Área externa cubierta para entrada de ambulancias;
5.2.2 Sala de recepción y espera, con sanitarios para usuarios;
5.2.3 Área de archivo de Historias Clínicas;
5.2.4 Área de clasificación de riesgo;
5.2.5 Área de higienización;
5.2.6 Consultorios;
5.2.7 Área para servicio social;
5.2.8 Sala de procedimientos con áreas delimitadas para sutura, curación, hidratación, y administración de medicamentos;
5.2.8.1 Estas áreas deberán estar separadas unas de otras por un medio físico;
5.2.9 Área para nebulización;
5.2.10 Sala para reanimación y estabilización;
5.2.11 Salas de observación y aislamientos;
5.2.12 Puesto de enfermería;
5.2.13 Sanitarios y duchas;
5.2.14 Depósito para residuos sólidos;
5.2.15 Depósito para material de limpieza;
5.2.16 Vestuarios y sanitarios para profesionales;
5.2.17 Farmacia;
5.2.18 Depósito de equipamientos e insumos.
5.3 Los servicios de urgencia y emergencia que prestan atención
quirúrgica deben contar en su área física o en el establecimiento donde
estuviese inserto, con:
5.3.1 Centro quirúrgico.
5.3.2 Áreas de apoyo técnico y logístico.
5.4 El servicio de urgencia y emergencia que presta atención
traumatológica y ortopédica debe contar en su área física o en el
establecimiento donde esté inserto, con sala para reducir fracturas y
enyesar.
5.5 El servicio de urgencia y emergencia debe poseer en sus instalaciones:
5.5.1 sistema de energía eléctrica de emergencia para los equipamientos
de soporte vital y los circuitos de iluminación de urgencia;
5.5.2 circuitos de iluminación distintos, de forma de evitar
interferencias electromagnéticas en el equipamiento y las instalaciones;
5.5.3 sistema de abastecimiento de gas medicinal, con punto de oxígeno,
y aire medicinal en las salas de nebulización, sala de observación y
sala de reanimación y estabilización.
5.6 El servicio de urgencia y emergencia debe poseer circulación y
puertas dimensionadas para el acceso de camillas y sillas de ruedas.
6. MATERIALES Y EQUIPAMIENTOS
6.1 El servicio de urgencia y emergencia debe mantener disponible en la unidad:
6.1.1 estetoscopio adulto e infantil;
6.1.2 esfigmomanómetro adulto e infantil;
6.1.3 otoscopio adulto e infantil;
6.1.4 oftalmoscopio;
6.1.5 espejo laríngeo;
6.1.6 resucitador manual con reservorio adulto e infantil;
6.1.7 desfibrilador
6.1.8. marcapasos externo;
6.1.9 monitor cardíaco;
6.1.10 oxímetro de pulso;
6.1.11 electrocardiógrafos;
6.1.12 equipamientos para detección de glucemia capilar;
6.1.13 aspiradores;
6.1.14 bombas de infusión con batería y equipo universal;
6.1.15 cilindro de oxígeno portátil y red canalizada de gases, definido de acuerdo al porte de la unidad;
6.1.16 camillas con ruedas y barandas;
6.1.17 máscara para resucitador adulto e infantil;
6.1.18 respirador mecánico adulto e infantil;
6.1.19 foco quirúrgico portátil;
6.1.20 foco quirúrgico con batería;
6.1.21 negatoscopio;
6.1.22 máscaras, sondas, drenajes, cánulas, pinzas y catéteres para diferentes usos;
6.1.23 laringoscopio adulto e infantil;
6.1.24 material para cricotiroidostomía;
6.1.25 equipos de macro y microgoteros;
6.1.26 material de cirugía menor;
6.1.27 collares de inmovilización cervical tamaños P, M y G;
6.1.28 plancha larga para inmovilización de la víctima en caso de trauma;
6.1.29 plancha corta para masaje cardíaco;
6.1.30 instrumentos necesarios para resucitación cardiorrespiratoria;
6.1.31 medicamentos que garanticen la asistencia en urgencias y emergencias;
6.1.32 asiento removible destinado al acompañante.
6.2 El servicio de urgencia y emergencia debe:
6.2.1 mantener instrucciones escritas, de uso y mantenimiento,
referentes a equipamientos o instrumentos, las que pueden ser
sustituidas o complementadas por manuales del fabricante;
6.2.2 asegurar el estado de integridad del equipamiento;
6.2.3 registrar la realización de los mantenimientos preventivos y correctivos.
6.3 los medicamentos, materiales, equipamientos e instrumentos
utilizados, nacionales e importados, deben estar regularizados de
acuerdo con la normativa nacional vigente.
7. ACCESO A LOS RECURSOS ASISTENCIALES
7.1 El servicio de urgencia y emergencia debe disponer o garantizar el
acceso, en el tiempo debido, a los siguientes recursos asistenciales,
diagnósticos y terapéuticos, específicos para la franja etaria asistida:
7.1.1 cirugía general;
7.1.2 clínica y cirugía obstétrica y ginecológica;
7.1.3 clínica y cirugía vascular;
7.1.4 clínica y cirugía neurológica;
7.1.5 clínica y cirugía ortopédica y traumatológica;
7.1.6 clínica y cirugía oftalmológica;
7.1.7 clínica y cirugía urológica;
7.1.8 clínica y cirugía odontológica y buco maxilofacial;
7.1.9 clínica gastroenterológica;
7.1.10 clínica nefrológica;
7.1.11 clínica psiquiátrica;
7.1.12 clínica para quemados;
7.1.13 terapia intensiva;
7.1.14 radiología intervencionista;
7.1.15 nutrición, incluyendo nutrición enteral y parenteral;
7.1.16 hemoterapia;
7.1.17 diálisis;
7.1.18 laboratorio clínico, incluyendo microbiología y hemogasometría;
7.1.19 anatomía patológica;
7.1.20 radiología convencional, incluyendo aparato de radiografía móvil;
7.1.21 ultra-sonografía, inclusive portátil;
7.1.22 ecodoppler;
7.1.23 tomografía computarizada;
7.1.24 resonancia magnética;
7.1.25 fibrobroncoscopía;
7.1.26 endoscopía digestiva;
7.1.27 electroencefalografía.
8. PROCESOS OPERACIONALES ASISTENCIALES
8.1 El servicio de urgencia y emergencia debe prestar al paciente asistencia integral e interdisciplinaria cuando sea necesario.
8.2 El servicio de urgencia y emergencia debe realizar la clasificación de los pacientes por niveles de riesgos.
8.2.1 La clasificación de riesgo debe ser efectuada por profesionales de salud capacitados.
8.2.2 La clasificación de riesgo debe considerar el grado de necesidad
del paciente y el orden de atención debe darse de acuerdo con los
protocolos clínicos de servicio.
8.3 El servicio de urgencia y emergencia debe garantizar que la
transferencia del paciente, en caso de necesidad, sea realizada después
de asegurar la disponibilidad de camas en el servicio de referencia, en
transporte adecuado a las necesidades.
8.3.1 Cuando fuera necesario la transferencia para una Unidad de
Terapia Intensiva, ésta debe ser efectuada lo más rápido posible.
8.4 Un equipo de servicio de urgencia y emergencia debe:
8.4.1 implantar e implementar acciones de farmacovigilancia,
tecnovigilancia, hemovigilancia y acciones de prevención y control de
infecciones y de eventos adversos;
8.4.2 contribuir con la investigación epidemiológica de brotes y eventos adversos y adoptar medidas de control;
8.4.3 proceder al uso racional de medicamentos, especialmente de antimicrobianos.
8.5 Todo paciente debe ser evaluado por el equipo asistencial en todos
los turnos, con registro en la historia clínica legible y debidamente
rubricada.
9. TRANSPORTE INTERHOSPITALARIO
9.1 El servicio de urgencia y emergencia debe tener disponible, para el
transporte de pacientes, materiales y medicamentos de acuerdo a las
necesidades de atención.
9.2 Todo paciente grave debe ser transportado con acompañamiento
continuo de médico y personal de enfermería, con habilidad comprobada
para la atención de urgencia y emergencia, inclusive cardiorespiratoria.
9.3 El transporte del paciente debe ser realizado de acuerdo al manual
de normas, rutinas y procedimientos establecidos por el equipo del
servicio de forma de garantizar la continuidad de la asistencia.
10. BIOSEGURIDAD
10.1 El servicio de urgencia y emergencia debe mantener actualizadas y
disponibles, para todos los profesionales de la salud, instrucciones
escritas de bioseguridad.
MERCOSUR/GMC/RES. Nº 03/15
REQUISITOS DE BUENAS PRÁCTICAS EN PROCEDIMIENTOS PARA ORGANIZACIÓN Y FUNCIONAMIENTO DE LOS SERVICIOS DE TRASPLANTE DE ÓRGANOS
VISTO: El Tratado de Asunción y el Protocolo de Ouro Preto.
CONSIDERANDO:
Que es necesario contar con Requisitos de Buenas Prácticas en
procedimientos para organización y funcionamiento de los servicios de
trasplante de órganos.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar los Requisitos de Buenas Prácticas en procedimientos
para organización y funcionamiento de los servicios de trasplante de
órganos, que constan como Anexo y forman parte de la presente
Resolución.
Art. 2 - Los Requisitos de Buenas Prácticas establecidos en la presente
Resolución deben ser incluidos en las normas de organización y
funcionamiento de los servicios de trasplante de órganos de cada Estado
Parte, pudiendo agregarse otros requisitos a la normativa nacional o
local de acuerdo con la necesidad de cada Estado Parte.
Art. 3 - Los Estados Partes indicarán, en el ámbito del SGT N° 11, los
organismos nacionales competentes para la implementación de la presente
Resolución.
Art. 4 - Esta Resolución deberá ser incorporada al ordenamiento jurídico de los Estados Partes, antes del 1/XII/2015.
XCVIII GMC - Brasilia, 29/V/15.
ANEXO
REQUISITOS DE BUENAS PRÁCTICAS EN PROCEDIMIENTOS PARA ORGANIZACIÓN Y FUNCIONAMIENTO DE LOS SERVICIOS DE TRASPLANTE DE ÓRGANOS
1. OBJETIVO
Establecer los Requisitos de Buenas Prácticas en procedimientos para
organización y funcionamiento de los servicios de trasplante de órganos.
2. DEFINICIÓN
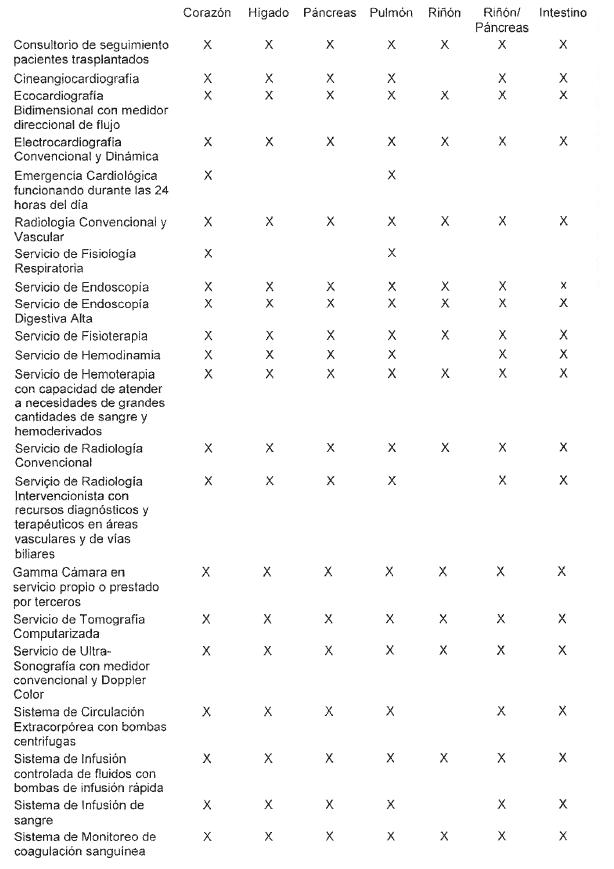
Programas de trasplante de órganos (corazón, pulmón, hígado, páncreas,
intestino y riñón): son aquellos destinados a la atención de pacientes
en lista de espera para trasplante y en seguimiento post trasplante, y
por lo tanto requieren instituciones que posean infraestructura, equipo
de profesionales específicamente capacitado, equipamiento específico y
otras tecnologías necesarias al diagnóstico y tratamiento.
3. REQUISITOS
3.1 Documentar y cumplir con los requisitos legales y principios éticos y de calidad establecidos por la OMS.
3.2 Todo programa de trasplante de órganos, público o privado, debe
estar inserto en un establecimiento de salud que tenga Habilitación o
Licencia de Funcionamiento, expedida por el órgano sanitario competente.
3.3 Toda institución con programa de trasplante deberá implementar las
acciones necesarias a fin de asegurar el funcionamiento de una unidad
de procuración de órganos y tejidos en la misma con actividad constante.
3.4 La construcción, reforma o adaptación en la estructura física de
las áreas destinadas al trasplante o la internación del paciente en el
post trasplante debe estar precedida de la aprobación del proyecto por
el órgano sanitario competente.
3.5 Es de responsabilidad de la administración del establecimiento de
salud prever y proveer los recursos humanos, equipamientos, materiales
y medicamentos necesarios a la operacionalización de los Programas de
Trasplantes de órganos.
3.6 La dirección del establecimiento de salud y el Jefe del equipo de
trasplante tienen la responsabilidad de planear, implementar y
garantizar la calidad de los procesos.
3.7 Cada programa de trasplante debe disponer de instrucciones escritas y actualizadas de las rutinas técnicas implantadas.
3.8 Las rutinas técnicas deben ser elaboradas en conjunto con los
servicios involucrados en la asistencia al paciente, asegurando la
asistencia integral y la interdisciplinariedad.
3.9 Un programa de trasplante de órganos debe:
3.9.1 poseer una estructura organizacional documentada.
3.9.2 preservar la identidad y la privacidad del paciente, asegurando un ambiente de respeto y dignidad.
3.9.3 promover ambiente acogedor.
3.9.4 incentivar y promover la participación de la familia en la atención al paciente crítico.
3.9.5 proveer orientaciones a los familiares en un lenguaje claro,
sobre el estado de salud del paciente y la asistencia a ser brindada,
desde la admisión hasta el alta.
4. RECURSOS HUMANOS
4.1 Todo programa de trasplante de órganos debe disponer del siguiente equipo:
4.1.1 Un jefe y subjefe de equipo médico, legalmente habilitados como
especialistas en la especialidad quirúrgica o clínica correspondiente
al órgano a trasplantar, específico para la modalidad de asistencia
adulto o pediátrico.
4.1.2 El médico a cargo de la jefatura y subjefatura del programa de
trasplante solo puede asumir la responsabilidad por un solo programa de
trasplante pudiendo ser integrante de otros programas de trasplante del
mismo órgano.
4.1.3 El jefe o subjefe de equipo podrán serlo de más de un programa de
trasplante de órganos cuando los mismos correspondan a órganos
diferentes.
4.1.4 En caso de ausencia del jefe de equipo, el subjefe será el profesional legalmente habilitado para sustituirlo.
4.1.5 El equipo de profesionales de un programa de trasplante de
órganos deberá asegurar la evaluación pre trasplante y el seguimiento
posterior al mismo de los pacientes bajo su atención.
4.1.6 El equipo de profesionales deberá estar conformado
preferentemente por dos médicos con especialidad quirúrgica y dos
profesionales clínicos, en ambos casos con especialidad del órgano a
trasplantar.
4.2 El jefe de equipo será responsable de implementar y mantener
registros de programa de educación permanente para todo el equipo de
profesionales del programa de trasplante que dirige.
4.3 La capacitación de experiencia quirúrgica y clínica en trasplante
de los integrantes del equipo de trasplante tuvo que haber sido
adquirida dentro de los últimos cinco (5) años respecto de la fecha en
la que se solicite la autorización para dicha práctica ante la
autoridad sanitaria competente.
4.4 Serán obligaciones del jefe de equipo:
4.4.1 Cumplir y hacer cumplir las normas y actos administrativos
vinculados con las leyes vigentes que regulen la actividad de donación
y trasplante en el país que realicen la mencionada práctica, como así
también las disposiciones de carácter administrativo emanadas de la
dirección del establecimiento asistencial en el cual realicen las
prácticas médico-quirúrgicas.
4.4.2 Cumplir en tiempo y forma con las disposiciones relativas a los
registros, protocolos e informes médicos y estadísticos contemplados en
las normas citadas en el punto precedente y en otras resoluciones o
disposiciones dictadas en la materia; asegurando dicha información a
los Organismos Nacionales de Donación y Trasplante, garantizando la
adecuada información a los pacientes.
4.4.3 Coordinar las acciones de los integrantes del equipo a su cargo a
los fines del estricto cumplimiento de las leyes vigentes garantizando;
4.4.3.1 la operatividad del mismo las veinticuatro (24) hs. de los trescientos sesenta y cinco (365) días del año;
4.4.3.2 la calidad de la atención a los pacientes inscriptos en lista de espera para trasplante o trasplantados.
4.4.4 Proponer las modificaciones de su equipo en el momento que lo
considere necesario, debiendo informar dentro de las cuarenta y ocho
(48) hs. por medio fehaciente las bajas o nuevas incorporaciones de sus
integrantes, a fin de ser tramitadas como nuevas autorizaciones.
4.4.5 Documentar la aceptación o no, del paciente al cambio de centro
de trasplante por falta de operatividad del equipo, la que deberá ser
notificada al Organismo Nacional de Donación y Trasplante competente,
siendo solidariamente responsable de su cumplimiento el director
técnico/médico del establecimiento.
5. INFRAESTRUCTURA FÍSICA Y EQUIPAMIENTO
5.1 Toda institución solicitante deberá encontrarse previamente
habilitada como establecimiento asistencial por la autoridad sanitaria
correspondiente.
5.2 Requisitos a cumplir por los establecimientos públicos o privados
que cuentan con servicios destinados a la ablación e implante de
órganos:
5.2.1 Quirófano adecuado y disponible a los fines del trasplante de
órganos, con instrumental quirúrgico acorde y suficiente, equipo de
monitoreo, cardioversión, estimulación eléctrica cardíaca y perfusión
vascular.
5.2.2 Las instituciones en donde funcionen programas de trasplante de órganos deberán contar con los siguientes servicios:
5.3 Los servicios prestados por terceros deben estar formalizados a
través de un contrato y cumplir con la normativa nacional vigente.
5.3.1 Deberá contar con dos (2) áreas de internación:
5.3.1.1 Unidad de cuidados intensivos, destinada a pacientes en el
postoperatorio inmediato o en situación de complicación y riesgo,
aislados dentro del sector que corresponda.
5.3.1.2 Área de internación clínica, con aislamientos adecuados a la situación requerida por el cuadro clínico del paciente.
5.4 Se considerarán como servicio aquellas áreas, sectores o unidades
operativas que representen el conjunto de recursos humanos
(profesionales, técnicos y administrativos), tecnológicos, de
equipamiento y de infraestructura que, organizados adecuadamente,
permitan el funcionamiento regular y permanente del mismo, garantizando
la resolución de los casos y procedimientos médicos que se presenten,
cualquiera sea su complejidad.
5.5 Los programas de trasplante de órganos deben disponer de
infraestructura física con ambientes e instalaciones necesarios para la
asistencia y la realización de los procedimientos con seguridad y
calidad.
6. REQUISITOS Y CRITERIOS PARA LA RENOVACIÓN DE LA AUTORIZACIÓN/HABILITACIÓN
6.1 Los periodos de renovación de la autorización/habilitación deberán
estar en conformidad con la legislación vigente en cada país.
6.2 A los efectos de proceder a la renovación de la
autorización/habilitación de establecimientos y reacreditación de
jefes, subjefes e integrantes de equipos, se deberán cumplimentar los
siguientes requisitos:
6.2.1 Nueva inspección satisfactoria de la infraestructura asistencial y evaluación de la actividad de la unidad de procuración.
6.2.2 Cumplimiento de los requisitos legales y normativos referente a cantidad y tipo de recursos humanos.
6.2.3 Cumplimiento efectivo de los requerimientos de información solicitados por la autoridad sanitaria competente.
6.2.4 Cumplimiento por parte del Director del establecimiento y del
jefe de equipo de trasplante, de la remisión al Organismo Nacional de
Donación y Trasplante que corresponda, de toda la información referida
a inscripción y baja de pacientes en lista de espera, como así también
al trasplante y seguimiento posterior, la cual deberá ser consignada en
los protocolos correspondientes.
6.3 Los programas que soliciten rehabilitación deberán certificar haber
efectuado actividad de trasplante con donante vivo y/o cadavérico
durante los dos (2) años previos a la misma.
6.4 La evaluación de los resultados de cada programa de acuerdo a
estándares nacionales que cada país a través de la Autoridad competente
establecerá y que tendrán en cuenta:
6.4.1 el número de trasplantes efectuados;
6.4.2 la relación ofertas/rechazos de órganos para trasplante;
6.4.3 la sobrevida del injerto y del paciente.
MERCOSUR/GMC/RES. Nº 04/15
PLAN REGIONAL DE SALUD Y SEGURIDAD DE LOS TRABAJADORES EN EL MERCOSUR
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto, el Acuerdo
Multilateral de Seguridad Social del Mercado Común del Sur, el Acuerdo
sobre Residencia para Nacionales de los Estados Partes del MERCOSUR, la
Declaración Sociolaboral del MERCOSUR y la Decisión Nº 64/10 del
Consejo del Mercado Común.
CONSIDERANDO:
Que la finalidad del Tratado de Asunción es unir cada vez más a los
pueblos de los Estados Partes, buscando caminos de integración y
progreso económico y social, para todos los habitantes del MERCOSUR por
lo tanto, su dimensión social se constituye en el lugar natural para
desarrollar las políticas orientadas a la justicia social e inclusión,
en beneficio de sus nacionales.
Que en este proceso de desarrollo económico con justicia social es
clave la decisión de avanzar en medidas para promover y proteger la
salud y seguridad de los trabajadores en el MERCOSUR.
Que el Artículo 17° de la Declaración Sociolaboral del MERCOSUR
establece el derecho de que todo trabajador ejerza sus actividades en
un ambiente de trabajo sano y seguro, que preserve la salud física y
mental del trabajador y estimule su desarrollo y desempeño profesional.
Que asimismo el segundo párrafo del citado artículo dispone que “Los
Estados Partes se comprometen a formular, aplicar y actualizar, en
forma permanente y en cooperación con las organizaciones de empleadores
y de trabajadores, políticas y programas en materia de salud y
seguridad de los trabajadores y del medio ambiente del trabajo, con el
fin de prevenir los accidentes de trabajo y las enfermedades
profesionales promoviendo condiciones ambientales propicias para el
desarrollo de las actividades de los trabajadores.”
Que la salud y seguridad de los trabajadores además de ser un tema de
competencia especifica del Subgrupo de Trabajo Nº 10 Relaciones
Laborales, Empleo y Seguridad Social (SGT Nº 10), ha sido incorporada
al Plan de Acción para la conformación de un Estatuto de la Ciudadanía
del MERCOSUR y a los “Ejes, Directrices y Objetivos Prioritarios del
Plan Estratégico de Acción Social del MERCOSUR (PEAS) .
Que la implementación de un plan regional de salud y seguridad de los
trabajadores en el MERCOSUR permitirá abordar este tema desde la
especificidad propia de las relaciones laborales, propiciando el
diálogo social junto a los actores sociales del mundo del trabajo.
El GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar el “Plan Regional de Salud y Seguridad de los
Trabajadores en el MERCOSUR”, que consta como Anexo y forma parte de la
presente Resolución.
Art. 2 - Esta Resolución no necesita ser incorporada al ordenamiento
jurídico de los Estados Partes, por reglamentar aspectos de la
organización o del funcionamiento del MERCOSUR.
XCVIII GMC - Brasilia, 29/V/15.
ANEXO
PLAN REGIONAL DE SALUD Y SEGURIDAD DE LOS TRABAJADORES EN EL MERCOSUR
El presente Plan tiene como objetivo general desarrollar acciones, de
aplicación progresiva, tendientes a promover y proteger la salud y
seguridad de los trabajadores en el MERCOSUR.
A tal fin, el plan se desarrolla sobre tres dimensiones temáticas: Normativa, Formativa y de Inspección.
Cada una tendrá un objetivo específico, para cuyo cumplimiento se prevé
la realización de acciones en el ámbito del Subgrupo de Trabajo Nº 10
Relaciones Laborales, Empleo y Seguridad Social.
Para la implementación del presente Plan el SGT N° 10 podrá contar con
una instancia de apoyo en el ámbito del SGT N° 10, a ser establecida de
conformidad con la normativa vigente.
El SGT N° 10 informará bianualmente al GMC de los avances alcanzados con relación a las acciones previstas en el presente Plan.
OBJETIVO GENERAL:
Desarrollar acciones tendientes a promover y proteger la salud y seguridad de los trabajadores en el MERCOSUR.
1.- DIMENSIÓN NORMATIVA
Objetivo específico:
Analizar la aplicación práctica del Artículo 17° de la Declaración
Sociolaboral del MERCOSUR y promover el diseño de políticas para
mejorar el goce de los derechos de salud y seguridad en el trabajo, por
parte de los trabajadores en el MERCOSUR.
Acciones:
- Promover el diseño de políticas para la aplicación de las
disposiciones del Artículo 17° de la Declaración Sociolaboral del
MERCOSUR.
- Realizar estudios de legislación comparada de los Estados Partes, en
materia de salud y seguridad en el trabajo y realizar propuestas de
carácter normativo.
- Colaborar junto con el Observatorio del Mercado de Trabajo del
MERCOSUR en el desarrollo de indicadores armonizados de entorno de
trabajo seguro para su implementación.
2.- DIMENSIÓN FORMATIVA
Objetivo específico:
Facilitar el conocimiento de los derechos y obligaciones en materia de salud y seguridad en el trabajo, en el MERCOSUR.
Acciones:
- Implementar acciones de información sobre derechos y obligaciones en materia de salud y seguridad en el trabajo.
- Fomentar actividades de formación para la implementación de sistemas
de gestión de salud y seguridad en los lugares y ambientes de trabajo.
- Desarrollar campañas de prevención para sectores de actividad determinados.
3.- DIMENSIÓN DE INSPECCIÓN
Objetivo específico:
Profundizar la inspección de las condiciones de salud y seguridad en el trabajo, en el MERCOSUR.
Acciones:
- Realizar operativos de inspección conjunta, bajo protocolos
armonizados, en sectores de actividad determinados, principalmente
aquellos con movimientos transfronterizos de trabajadores.
- Capacitar a los inspectores del trabajo en materia de salud y
seguridad en general y en particular para el cumplimiento de las
Decisiones CMC Nº 32/06 y 33/06.
- Cooperar en el logro de los objetivos del “Plan Regional de Inspección del Trabajo del MERCOSUR” (Resolución GMC N° 22/09).
MERCOSUR/GMC/RES. N° 05/15
MODIFICACIÓN DE LA NOMENCLATURA COMÚN DEL MERCOSUR Y SU CORRESPONDIENTE ARANCEL EXTERNO COMÚN
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto, las
Decisiones N° 07/94, 22/94 y 31/04 del Consejo del Mercado Común y la
Resolución Nº 05/11 del Grupo Mercado Común.
CONSIDERANDO:
Que se hace necesario ajustar la Nomenclatura Común del MERCOSUR y su
correspondiente Arancel Externo Común, instrumentos esenciales de la
Unión Aduanera.
EL GRUPO MERCADO COMÚN
RESUELVE:
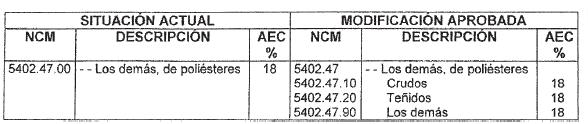
Art. 1 - Aprobar las modificaciones a la Nomenclatura Común del
MERCOSUR y su correspondiente Arancel Externo Común, que constan como
Anexo y forman parte de la presente Resolución.
Art. 2 - Las modificaciones de la Nomenclatura Común del MERCOSUR y su
correspondiente Arancel Externo Común, aprobadas por la presente
Resolución, tendrán vigencia antes del 1/I/2016, debiendo los Estados
Partes asegurar su incorporación a sus respectivos ordenamientos
jurídicos nacionales antes de esa fecha.
XCVIII GMC - Brasilia, 29/V/15.
ANEXO
MERCOSUR/GMC/RES. Nº 06/15
ADDENDUM N° 5 AL CONVENIO DE FINANCIACIÓN N° DCI-ALA/2008/020-297 MERCOSUR - UNIÓN EUROPEA PROGRAMA “MERCOSUR AUDIOVISUAL”
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto, la Decisión
N° 23/14 del Consejo del Mercado Común y las Resoluciones N° 27/09,
04/12, 02/14, 30/14 y 38/14 del Grupo Mercado Común.
CONSIDERANDO:
Que el 22 de julio de 2009 fue suscripto el Convenio de Financiación
entre el MERCOSUR y la Unión Europea, que tiene como objetivo
fortalecer el sector cinematográfico y audiovisual del MERCOSUR.
Que mediante la Decisión CMC Nº 23/14 se delegó en el Grupo Mercado
Común la facultad de suscribir convenios en el marco de la negociación
de programas de cooperación técnica.
Que resulta necesario postergar la finalización del período de ejecución del Programa.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar la suscripción del Addendum N° 5 al Convenio de
Financiación N° DCI- ALA/2008/020-297 MERCOSUR - Unión Europea Programa
“MERCOSUR Audiovisual”, que consta como Anexo y forma parte de la
presente Resolución.
Art. 2 - El Anexo de la presente Resolución se encuentra únicamente en idioma español.
Art. 3 - Esta Resolución no necesita ser incorporada al ordenamiento
jurídico de los Estados Partes, por reglamentar aspectos de la
organización o del funcionamiento del MERCOSUR.
XCVIII GMC - Brasilia, 29/V/15.
ADDENDUM Nº 5
AL CONVENIO DE FINANCIACIÓN ENTRE LA UNIÓN EUROPEA Y EL MERCOSUR
“MERCOSUR Audiovisual”
La Unión Europea, en lo sucesivo denominada “la UE”, representada por
la Comisión Europea, en lo sucesivo denominada “la Comisión”
por una parte, y
El MERCOSUR (Argentina, Brasil, Paraguay, Uruguay), representado por el
Grupo Mercado Común del MERCOSUR, en lo sucesivo “el Beneficiario”.
por otra,
HAN CONVENIDO EN LO SIGUIENTE:
ANEXO II
DISPOSICIONES TÉCNICAS Y ADMINISTRATIVAS (DTAs)
3. LOCALIZACIÓN Y DURACIÓN
DONDE DICE:
[…] 3.2. Duración
El período de ejecución del convenio será de 88 meses. Este período de
ejecución comprenderá dos fases, de acuerdo con las condiciones del
artículo 4.1 de las condiciones Generales (Anexo I del presente
Convenio):
• Fase de ejecución operativa, que comenzara a partir de la entrada en
vigor del convenio de financiación y tendrá una duración de 70 meses.
• Fase de cierre, de una duración de 18 meses, que comenzara a partir
de la fecha de vencimiento de la fase de ejecución operativa.
DEBE DECIR:
[…] 3.2. Duración
El período de ejecución del convenio será de 88 meses. Este período de
ejecución comprenderá dos fases, de acuerdo con las condiciones del
artículo 4.1 de las condiciones Generales (Anexo I del presente
Convenio):
• Fase de ejecución operativa, que comenzara a partir de la entrada en
vigor del convenio de financiación y tendrá una duración de 73 meses.
• Fase de cierre, de una duración de 15 meses, que comenzara a partir
de la fecha de vencimiento de la fase de ejecución operativa.
Todas las demás estipulaciones del convenio de financiación se mantienen sin cambios.
El presente addendum entrará en vigor en la fecha de la última firma de
las dos partes. Hecho en 6 ejemplares con valor de original en lengua
española, habiéndose entregado dos ejemplares a la Comisión y cuatro al
Beneficiario.
MERCOSUR/GMC/RES. Nº 07/15
DEROGACIÓN DE LAS RESOLUCIONES GMC Nº 97/94, 13/96, 22/96 Y 23/08
VISTO: El Tratado de Asunción, el Proto colo de Ouro Preto y las
Resoluciones Nº 97/94, 13/96, 22/96 y 23/08 del Grupo Mercado Común.
CONSIDERANDO:
Que conforme surge de la revisión periódica del acervo normativo del
MERCOSUR, resulta conveniente derogar aquellas normas que han cumplido
el mandato para el cual fueron aprobadas.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Derogar las Resoluciones GMC Nº 97/94, 13/96, 22/96 y 23/08.
Art. 2 - Esta Resolución necesita ser incorporada sólo al ordenamiento
jurídico interno de Argentina, Brasil, Paraguay y Uruguay. Esta
incorporación deberá ser realizada antes del 1/XII/2015.
XCVIII GMC - Brasilia, 29/V/15.
MERCOSUR/GMC/RES. Nº 08/15
DEROGACIÓN DE LAS RESOLUCIONES GMC Nº 13/08 Y 54/08
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto y las Resoluciones Nº 13/08 y 54/08 del Grupo Mercado Común.
CONSIDERANDO:
Que conforme surge de la revisión periódica del acervo normativo del
MERCOSUR, resulta conveniente derogar aquellas normas que han cumplido
el mandato para el cual fueron aprobadas.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Derogar las Resoluciones GMC Nº 13/08 y 54/08.
Art. 2 - Esta Resolución no necesita ser incorporada al ordenamiento
jurídico de los Estados Partes, por reglamentar aspectos de la
organización o del funcionamiento del MERCOSUR.
XCVIII GMC - Brasilia, 29/V/15.
MERCOSUR/GMC/RES. Nº 09/15
CERTIFICADO DE VENTA LIBRE DE PRODUCTOS DOMISANITARIOS
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto y las
Resoluciones N° 24/96, 25/96, 24/06 y 51/06 del Grupo Mercado Común.
CONSIDERANDO:
Que el Certificado de Venta Libre de Productos Domisanitarios permite
garantizar que los productos fabricados cumplen con la reglamentación
de la autoridad sanitaria de cada Estado Parte.
Que la emisión del Certificado de Venta Libre de Productos
Domisanitarios es competencia de la autoridad sanitaria de cada Estado
Parte.
Que es necesario armonizar los requisitos mínimos que deben ser
contemplados en el Certificado de Venta Libre de Productos
Domisanitarios.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar el contenido mínimo del modelo para “Certificado de
Venta Libre de Productos Domisanitarios”, que consta como Anexo y forma
parte de la presente Resolución.
Art. 2 - Los Estados Partes, por medio de las autoridades sanitarias
competentes, emitirán el Certificado de Venta Libre de Productos
Domisanitarios a solicitud del titular del registro.
Art. 3 - La emisión y validez del Certificado de Venta Libre de
Productos Domisanitarios estarán sujetas a la comprobación de que los
productos cumplan con la normativa MERCOSUR vigente.
Art. 4 - A los efectos de la implementación de la presente Resolución,
la República del Paraguay utilizará la denominación Certificado de
Libre Venta de Productos Domisanitarios (CLV), conforme a los
requisitos establecidos en el Anexo.
Art. 5 - Los organismos nacionales competentes para la implementación
de la presente Resolución son las autoridades sanitarias de los Estados
Partes.
Art. 6 - Esta Resolución deberá ser incorporada al ordenamiento jurídico de los Estados Partes, antes del 1/XII/2015.
XCVIII GMC - Brasilia, 29/V/15.
ANEXO
CERTIFICADO DE VENTA LIBRE DE PRODUCTOS DOMISANITARIOS
La [Autoridad Sanitaria Competente del Estado Parte] CERTIFICA que el
producto abajo detallado, cumple debidamente con la reglamentación
nacional, en conformidad con la normativa MERCOSUR vigente.
MERCOSUR/GMC/RES. Nº 10/15
REGLAMENTO TÉCNICO MERCOSUR DE BUENAS
PRÁCTICAS DE FABRICACIÓN PARA PRODUCTOS DOMISANITARIOS (DEROGACIÓN DE
LAS RES. GMC Nº 56/96, 23/01 Y 09/04)
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto y las
Resoluciones Nº 24/96, 56/96, 38/98, 03/99, 23/01, 56/02 y 09/04 del
Grupo Mercado Común.
CONSIDERANDO:
Que los productos domisanitarios deben ser seguros bajo las condiciones normales y previsibles de uso.
Que la fiscalización de los establecimientos fabricantes e importadores
de productos domisanitarios, a través de inspecciones técnicas, es un
mecanismo que contribuye a garantizar la calidad con que llegan al
mercado los productos que elaboran, envasan e importan esos
establecimientos.
Que dicha fiscalización debe cubrir aspectos relativos a condiciones de
funcionamiento y sistemas de control de calidad utilizados por dichos
establecimientos.
Que existe la necesidad de establecer procedimientos comunes a ser
aplicados en los Estados Partes, usando uniformidad de criterios para
la evaluación de los establecimientos de fabricantes e importadores de
productos domisanitarios.
Que las acciones de control son responsabilidad de las autoridades
sanitarias competentes, quienes deben contar con un modelo que asegure
el control de las industrias con uniformidad de criterio y neutralidad,
simetría y reciprocidad en el tratamiento y aplicación de las normas de
regulación.
Que las Buenas Prácticas de Fabricación (BPF) deben reflejar los
requisitos mínimos indispensables a ser cumplidos por las industrias en
la fabricación, envasado, almacenamiento y control de calidad de los
referidos productos.
Que, como consecuencia de los avances tecnológicos y del carácter
dinámico de la reglamentación sanitaria, es necesario actualizar y
adoptar nuevas directrices sobre Buenas Prácticas de Fabricación con el
fin de garantizar la seguridad y calidad de los productos
domisanitarios.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar el “Reglamento Técnico MERCOSUR de Buenas Prácticas de
Fabricación para Productos Domisanitarios”, que consta como Anexo y
forma parte de la presente Resolución.
Art. 2 - Los organismos nacionales competentes para la implementación
de la presente Resolución son las autoridades sanitarias de los Estados
Partes.
Art. 3 - La presente Resolución será aplicada en el territorio de los
Estados Partes, al comercio entre ellos y a las importaciones extrazona.
Art. 4 - Derogar las Resoluciones GMC Nº 56/96, 23/01 y 09/04.
Art. 5 - Esta Resolución deberá ser incorporada al ordenamiento jurídico de los Estados Partes, antes del 10/XII/2015.
XCVIII GMC - Brasilia, 29/V/15.
ANEXO
REGLAMENTO TÉCNICO MERCOSUR DE BUENAS PRÁCTICAS PARA FABRICACIÓN DE PRODUCTOS DOMISANITARIOS
CONTENIDO
1. Consideraciones Generales
2. Definiciones
3. Gestión de Calidad
4. Requisitos básicos de Buenas Prácticas de Fabricación (BPF)
5. Salud, Sanitización, Higiene, Vestuario y Conducta
6. Reclamos
7. Retiro de Productos
8. Devolución
9. Autoinspección/ Auditoría Interna
10. Documentación y Registros
11. Personal
12. Instalaciones
13. Sistemas e Instalaciones de agua
14. Áreas Auxiliares
15. Recepción y Almacenamiento
16. Muestreo de Materiales
17. Producción/Elaboración
18. Control de calidad
19. Contramuestras
1. CONSIDERACIONES GENERALES
El objetivo de este Reglamento Técnico es el de reglamentar la
fabricación de productos domisanitarios, de modo que los factores
humanos, técnicos y administrativos (de la fabricación) que puedan
tener influencia en la calidad de los mismos sean eficazmente
controlados, teniendo como objetivo prevenir, reducir y eliminar
cualquier deficiencia en la calidad de los mismos, que pueden afectar
negativamente a la salud y seguridad del usuario.
Como consecuencia, este Reglamento Técnico reúne los elementos básicos
a considerar por cada empresa fabricante, de forma que pueda elaborar
eficazmente productos domisanitarios, garantizando al mismo tiempo la
seguridad del usuario y la conformidad de sus productos a los propios
patrones de calidad previamente establecidos y planificados, como
también los aspectos de seguridad e higiene relacionados con la
actividad.
Las Buenas Prácticas de Fabricación (BPF) son aplicables a todas las operaciones involucradas en la fabricación de productos.
Los aspectos de seguridad para el personal involucrado en la
fabricación y de protección ambiental están reglamentados por
legislación específica y los establecimientos deben cumplir con los
requisitos aplicables a cada una de las áreas.
2. DEFINICIONES
A los fines de este Reglamento Técnico se entiende por:
Acción Correctiva: acción adoptada para eliminar la causa de una no conformidad detectada u otra situación indeseable.
Acción Preventiva: acción adoptada para eliminar la causa de una potencial no conformidad u otra potencial situación indeseable.
Acondicionamiento: conjunto de operaciones a las que es sometido un
producto en su envase primario hasta la obtención de este mismo en su
presentación final.
Aprobado: condición en que se encuentran los materiales, insumos o productos que cumplen con las especificaciones establecidas.
Área dedicada: sector marcado o delimitado de uso exclusivo para una determinada actividad o proceso.
Area definida: sector marcado o delimitado para la realización de alguna actividad específica.
Área separada: sector delimitado físicamente y que constituye un recinto por sí mismo.
Área segregada: instalaciones que ofrecen separación completa y total
de todos los aspectos de una operación, incluyendo movimiento del
personal y equipamiento con procedimientos y controles bien
establecidos.
Auditoria: análisis sistemático e independiente que permite determinar
si las actividades de calidad y sus resultados cumplen con los
requisitos planificados y si tales requisitos fueron puestos en
práctica de manera efectiva. Debe ser realizado por personal calificado.
Auditoria externa: cuando la auditoría sea realizada por personas calificadas externas a la empresa.
Auditoria interna/Autoinspección: cuando la auditoría sea realizada por personal competente de la propia empresa.
Buenas Prácticas de Fabricación: requisitos generales que el fabricante
de producto debe aplicar a las operaciones de fabricación de productos
domisanitarios de modo de garantizar la calidad y seguridad de los
mismos.
Calibración: conjunto de operaciones de verificación y ajuste de
instrumentos o sistemas de medición según normas reconocidas, para que
funcionen dentro de sus límites de precisión y exactitud.
Calificación: conjunto de acciones realizadas para probar y documentar
que cualquiera de las instalaciones, sistemas o equipamientos estén
adecuados, instalados y/o funcionan correctamente y llevan a los
resultados esperados.
Contaminación: introducción indeseada de impurezas de naturaleza
física, química y/o microbiológica en la materia prima, material de
embalaje, producto intermedio y/o producto terminado durante la
fabricación.
Contaminación cruzada: contaminación de una materia prima, producto
intermedio o terminado con otra materia prima, producto intermedio o
terminado durante la fabricación.
Contramuestra: muestra de materia prima, materiales o producto
terminado, mantenida por el fabricante, debidamente identificada por un
período definido.
Control de Calidad: operaciones usadas para verificar el cumplimento de
los requisitos técnicos de acuerdo con las especificaciones previamente
definidas.
Control en proceso: verificaciones realizadas durante la elaboración
para monitorear y, si es necesario, ajustar el proceso para asegurar
que el producto cumpla con sus especificaciones.
Cuarentena: retención temporaria de materia prima, material de
embalaje, productos intermedios, semi-terminado, a granel o terminados,
en cuanto aguardan decisión de liberación, rechazo o reprocesamiento.
Envasado: todas las operaciones por las cuales el producto a granel
debe pasar, a fin de tornarse producto terminado, incluyendo
fraccionamiento, rotulado, y acondicionamiento cuando fuera el caso.
EPC: Equipamiento de Protección Colectiva.
EPI: Equipamiento de Protección Individual.
Especificación: documento que describe en detalles los requisitos que
deben cumplir los productos o materiales usados u obtenidos durante la
fabricación.
Establecimiento: unidades de la empresa donde se realizan actividades previstas por legislación sanitaria vigente.
Fabricación: todas las operaciones que incluyen la adquisición de
materiales, producción, control de calidad, liberación, almacenamiento,
expedición de productos terminados y los controles relacionados.
Fórmula de Fabricación/Orden de Producción: documento de referencia
para la producción de un lote que contemple las informaciones de la
fórmula patrón.
Fórmula Patrón: documento o grupo de documentos que especifican las
materias primas con sus cantidades y los materiales de embalaje,
juntamente con la descripción de los procedimientos y precauciones
necesarias para la elaboración de determinadas cantidades de productos
terminados. Además de eso, provee instrucciones sobre la elaboración
del producto, inclusive sobre los controles en proceso.
Fraccionamiento: operación que permite que el producto a granel, por medio de un proceso definido, sea envasado.
Garantía de Calidad: todas las acciones planeadas sistemáticas
necesarias para garantizar que un producto o servicio satisfacerá todos
los requisitos de calidad y seguridad en su uso previamente establecido.
Gestión de Calidad: actividades coordinadas para dirigir y controlar una organización, en lo que respecta a calidad.
Inspección: actividades tales como medición, ensayo, examen, de una o
más características de una entidad, producto o servicio, comparando los
resultados con requisitos específicos para establecer si la conformidad
de una característica es atendida.
Lote: cantidad definida de materia prima, material de embalaje o
producto terminado fabricado en un único proceso o serie de procesos,
cuyas características esenciales son la homogeneidad y calidad dentro
los límites especificados. En la fabricación continua, el lote
corresponde a una fracción definida de la producción.
Materia Prima: cualquier sustancia involucrada en la obtención de un
producto que sea parte de este en su forma original o modificada.
Material de Envase: cada uno de los elementos de acondicionamiento que
estarán en el producto final. Conforme entren o no en contacto con el
producto, se dividen en “primarios” o “secundarios”, respectivamente.
Muestreo: conjunto de operaciones de retiro y preparación de las muestras.
Número de Lote: referencia numérica, alfabética o alfanumérica o señal
que identifica específicamente un lote de materia prima, de material de
embalaje, producto obtenido por una operación u operaciones.
Procedimiento Operativo Estandarizado: procedimiento escrito y aprobado
que provee instrucciones detalladas para la realización de actividades
específicas.
Producción: todas las operaciones involucradas en la preparación de
determinado producto desde la recepción de materiales del
almacenamiento, pasando por el procesamiento y embalaje, hasta la
obtención del producto terminado.
Producción en campaña: elaboración secuencial de diversos lotes de un mismo producto.
Producto a granel: cualquier producto que ha pasado por todas las etapas de producción, sin incluir el proceso de envasado
Producto devuelto: producto que ya fue expedido y que retorna al fabricante o importador.
Producto retirado: producto expedido que retorna al fabricante o
importador por iniciativa propia y/o determinación de Autoridad
Sanitaria competente.
Producto Semi-elaborado/intermedio: sustancia o mezcla de sustancias
que requieran posteriores procesos de producción a fin de convertirse
en productos a granel.
Producto semi-terminado: producto que necesita de por lo menos una
operación posterior antes de ser considerado producto terminado.
Producto terminado: producto que haya pasado por todas las etapas de
producción y acondicionamiento, listo para la venta y su uso.
Plan de validación: documento que describe las actividades a ser realizadas en la validación.
Reanálisis: ensayo realizado en materiales, previamente aprobados, para
confirmar el mantenimiento de las especificaciones establecidas, dentro
de su plazo de validez.
Reclamo: notificación externa que puede ser indicativo de posibles desvíos de calidad.
Registro de Lote: toda documentación relativa a un lote de un producto específico.
Reprocesamiento: repetición de una o más etapas que ya forman parte del
proceso de fabricación establecido en un lote de producto que no cumple
con las especificaciones.
Retiro de producto: procedimiento ejecutado por la empresa de retirar
un producto del mercado. Puede ser espontáneo o determinado por la
autoridad sanitaria competente.
Sanitización: proceso utilizado para reducir el número de
microorganismos viables para niveles aceptables en una superficie
limpia.
Sistema de garantía de Calidad: conjunto de procedimientos para
obtención y mantenimiento de la calidad deseada, involucrando:
Planeamiento; Recursos; Instalaciones; Control de Proyecto;
Adquisiciones; Manufactura; Embalaje; Etiquetado; Almacenamiento;
Asistencia Técnica, de modo de asegurar la calidad, seguridad y
eficacia de los servicios y productos.
Tercerización: contratación de fabricación por terceros para la
ejecución de etapas parciales o totales relativas a la fabricación,
control de calidad o almacenamiento de productos domisanitarios.
Validación: acción documentada, conducida para establecer y demostrar
que un proceso o procedimiento conduce necesariamente y efectivamente
al objetivo requerido.
3. GESTIÓN DE CALIDAD
3.1 Los conceptos de Garantía de Calidad, de Buenas Prácticas de
Fabricación (BPF) y de Control de Calidad son aspectos
interrelacionados de la gestión de la calidad. Están descriptos en este
Reglamento de forma de enfatizar sus relaciones y su fundamental
importancia para la fabricación de los productos regidos por este
Reglamento.
3.2 Principios
3.2.1 La calidad debe ser responsabilidad de todo el personal de la
empresa teniendo como referencia la política de calidad, o sea, las
intenciones y directrices globales relativas a la calidad formalmente
expresada y autorizada por la dirección de la empresa.
3.2.2 La empresa debe establecer, documentar, implementar y mantener un
sistema eficaz y eficiente para la gestión de la calidad, con la
participación activa de todo el personal involucrado en la fabricación.
3.2.3 El sistema para la gestión de la calidad debe comprender la
estructura organizacional, los procedimientos, los procesos, los
recursos, la documentación y las actividades necesarias para asegurar
que el producto esté en conformidad con las especificaciones
pretendidas de calidad.
3.2.4 Todas las actividades relacionadas al sistema de la calidad deben ser documentadas.
3.3 Garantía de calidad
3.3.1 El fabricante debe mantener un Sistema de Garantía de la Calidad
3.3.2 El fabricante debe asegurar la calidad, seguridad y eficacia de
los productos y sistemas de apoyo relacionados a la producción,
debiendo cumplir los requisitos establecidos en la legislación
sanitaria vigente.
3.3.3 El sistema de Garantía de la Calidad debe ser constituido por
personal competente y calificado, instalaciones y equipos adecuados,
compatible con las actividades desarrolladas.
3.3.4. Un sistema de Garantía de Calidad debe asegurar que:
a) las operaciones de producción y control estén claramente especificadas por escrito y las exigencias de BPF cumplidas;
b) las responsabilidades gerenciales estén claramente definidas y documentadas;
c) se relicen todos los controles establecidos como necesarios en las
materias primas, materiales de empaque, productos semielaborados,
productos a granel, productos semi-terminados, productos terminados, y
los relativos a control en proceso, calibraciones, calificaciones y
validaciones cuando aplique;
d) los productos no sean comercializados o entregados al consumo antes
que sean realizadas todas las etapas de control y liberación;
e) se provean instrucciones para garantizar que los productos sean
manipulados, almacenados y distribuidos de forma que la calidad de los
mismos sea mantenida por todo el plazo de validez;
f) exista un procedimiento de auto-inspección de la calidad que evalúe
regularmente la efectividad y la aplicación del Sistema de Garantía de
Calidad;
g) los desvíos de calidad, los eventos adversos y los reclamos sean
reportados, investigados, registrados y que sean implementadas las
acciones correctivas necesarias;
h) los procedimientos, especificaciones e instrucciones que puedan
tener influencia en la calidad de los productos sean periódicamente
revisados y mantenidos los respectivos historiales;
i) la estabilidad de un producto sea determinada conforme al reglamento
específico y que los estudios sean repetidos después de cualquier
cambio significativo en los procesos de producción, formulación,
equipamientos o materiales de embalaje;
j) la trazabilidad de todos los procesos relativos a la fabricación del producto sea garantizada;
3.3.5 Deben existir criterios definidos para la calificación de
proveedores la cual podrá incluir: evaluación del historial del
proveedor, la evaluación preliminar a través de cuestionario y/o
auditorias de calidad.
3.4 Validación
3.4.1. La empresa debe conocer sus procesos a fin de establecer
criterios para identificar la necesidad de validación o no de los
mismos. Cuando las validaciones fueran aplicables debe ser establecido
un protocolo de validación que especifique como el proceso será
conducido. El protocolo debe ser aprobado por Garantía de Calidad.
3.4.2 Para los productos/procesos que no serán validados, la empresa
debe establecer todos los controles operacionales necesarios para
garantizar el cumplimiento de los requisitos preestablecidos o
especificados.
3.4.3 El protocolo de validación debe especificar como mínimo:
a) descripción de los equipos e instalaciones;
b) variables a ser monitoreadas;
c) muestras que deben ser tomadas (ubicación, frecuencia, cantidad y método de muestreo);
d) características de desempeño a ser monitoreados, especificando los métodos analíticos y limites de aceptación;
e) definición de las responsabilidades;
f) descripción de los métodos utilizados para registro y evaluación de los resultados;
g) criterios de aceptación;
h) capacitación necesaria para el programa de validación.
3.4.4 Es recomendable la validación de la limpieza, metodología
analítica (cuando se trate de metodologías no codificadas en normas u
otra bibliografía reconocida), sistemas informatizados, sistema de agua
de procesos.
3.4.5 El informe de validación deberá hacer referencia al protocolo, y
ser elaborado contemplando resultados obtenidos, desvíos, conclusiones,
recomendaciones y cambios.
3.4.6 Cualquier desvío del protocolo de validación debe ser documentado, investigado y justificado.
3.4.7 El proceso de validación es satisfactorio cuando los resultados
son aceptables. Caso contrario se debe analizar el origen de los
desvíos encontrados y determinar las alteraciones necesarias, hasta que
el mismo presente resultados aceptables.
3.4.8 Deben ser establecidos los criterios de calificación de acuerdo
con la complejidad de los equipamientos, procesos y sistemas críticos.
La calificación es un prerrequisito para la validación.
3.5 Revalidación
3.5.1 En el caso de procesos y sistemas validados, la empresa debe
determinar la necesidad de su revalidación considerando el historial de
los resultados, verificando que el proceso es consistente con la última
validación.
3.5.2 Cada cambio debe ser evaluado por Garantía de Calidad, para
determinar la necesidad o no de revalidación, considerando el impacto
sobre los procesos y sistemas ya validados.
3.5.3 La extensión de la revalidación depende de la naturaleza de los
cambios y de cómo ellas afectan los diferentes aspectos de los procesos
y sistemas previamente validados.
3.5.4 La empresa debe definir la periodicidad de la revalidación.
3.6 Estabilidad
3.6.1 La empresa durante el proceso de desarrollo debe establecer
estudios de estabilidad de los productos contemplando los
procedimientos y registros con: resultados de los análisis,
metodologías analíticas, condiciones de conservación de la muestra,
periodicidad de análisis y fecha de vencimiento.
3.6.2 Se deben mantener registros de los análisis efectuados y de los estudios de estabilidad realizados.
4. REQUISITOS BÁSICOS DE BUENAS PRACTICAS DE FABRICACION (BPF)
4.1 Las BPF determinan que:
a) los procesos de fabricación deben ser claramente definidos,
sistemáticamente revisados, y mostrar que son capaces de fabricar
productos dentro de los patrones de calidad exigidos, atendiendo a las
respectivas especificaciones;
b) las etapas críticas de los procesos de fabricación y cualquier
modificaciones significativas deben ser sistemáticamente controladas y
cuando sea posible validarlas;
c) las áreas de fabricación deben estar provistas de la infraestructura
necesaria para realización de las actividades, incluyendo:
i Personal entrenado y calificado
ii Instalaciones y espacios adecuados
iii Servicios y equipamientos apropiados
iv Rótulos, embalajes y materiales apropiados
v Instrucciones y procedimientos aprobados
vi Depósitos apropiados
vii Personal, laboratorio y equipamientos adecuados para el control de calidad
d) las instrucciones y los procedimientos deben ser escritos en
lenguaje claro y objetivo y ser aplicable a las actividades realizadas;
e) el personal debe estar entrenado para desempeñar correctamente los procedimientos;
f) deberán realizarse registros durante la producción para demostrar
que todas las etapas constantes en los procedimientos e instrucciones
fueran seguidas y que la cantidad y la calidad del producto obtenido
están de conformidad con lo esperado. Cualquier desvios significativos
deben ser registrados, investigados y corregidos;
g) los registros referentes a la fabricación deben estar archivados de
manera organizada y de fácil acceso, permitiendo la trazabilidad;
h) el almacenamiento y la distribución interna de los productos deben minimizar cualquier riesgo que afecte su calidad;
i) se deberá implementar un procedimiento de retiro de cualquier lote después de su venta o provisión;
j) los reclamos sobre productos comercializados deben ser registrados y
examinados. Las causas de los desvíos de calidad deben ser investigadas
y documentadas. Deben ser tomadas medidas en relación a los productos
con desvío de calidad y adoptadas las medidas a fin de de prevenir
reincidencia.
5. SALUD, SANITIZACIÓN, HIGIENE, VESTUARIO Y CONDUCTA
5.1 Las actividades de sanitización e higiene deben comprender:
personal, instalaciones, equipamientos y utensilios, materiales de
producción y recipientes, productos para limpieza y desinfección y
cualquier otro aspecto que pueda constituir fuente de contaminación
para el producto. Las fuentes potenciales de contaminación deben ser
eliminadas a través de un adecuado programa de sanitización e higiene.
5.2 Todo el personal debe ser sometido a exámenes de salud para la
admisión y posteriormente a exámenes periódicos, necesarios para las
actividades desempeñadas, de acuerdo con procedimientos establecidos
por la empresa.
5.3. Todo el personal debe ser entrenado en las prácticas de higiene
personal. Todas las personas involucradas en los procesos de
fabricación deben cumplir con las normas de higiene personal conforme a
procedimientos internos de la empresa.
5.4 Las personas con sospecha o confirmación de enfermedades o lesiones
expuestas que puedan afectar de forma adversa la calidad de los
productos, no deben manipular materias primas, materiales de empaque,
productos semi-elaborados/semi-terminados, granel o productos
terminados hasta que su condición de salud no represente riesgo al
producto.
5.5 Todo el personal debe ser instruido e incentivado a informar a su
supervisor inmediato cualquier situación adversa, relativa a la
producción, al equipamiento o al personal, que consideren que puedan
interferir en los productos.
5.6. La empresa debe asegurar que las materias primas, materiales de
embalaje primarios, productos semi-terminados y a granel sean
manipulados de forma de garantizar la protección de los materiales
contra contaminaciones.
5.7 La empresa debe asegurar que los empleados utilicen indumentaria
limpia y apropiada para cada área y actividad para garantizar la
protección del producto contra contaminaciones.
5.8 Para que sea asegurada la protección del personal, el fabricante
debe disponer de Equipamiento de Protección Colectiva (EPC) y
Equipamiento de Protección Individual (EPI) de acuerdo con las
actividades desarrolladas conforme legislación específica.
5.9 Se prohíbe fumar, comer, beber o mascar, tener alimentos, bebidas,
cigarros, medicamentos personales y plantas en las áreas de producción,
del laboratorio de control de calidad y de almacenamiento o en
cualquier otras áreas en que tales acciones puedan influir adversamente
en la calidad del producto. La empresa debe garantizar la adecuada
comunicación de esta prohibición.
5.10 La localización de los bebederos debe estar restringida a
corredores o lugares específicos, de modo de evitar contaminación del
producto y/o riesgo a la salud del trabajador.
5.11 Visitantes y personas no entrenadas sólo podrán acceder a las
áreas de producción después de la orientación sobre normas de higiene y
seguridad, utilizando indumentaria adecuada y acompañadas por
profesional designado.
6. RECLAMOS
6.1 Los reclamos y demás informaciones referentes a productos con
posibles desvíos de calidad deben ser cuidadosamente investigados y
registrados de acuerdo con procedimientos escritos. La gestión de estas
investigaciones debe ser realizada por personal autorizado con la
participación de Control de Calidad y las demás áreas involucradas.
6.2 En caso que sea necesario, la verificación debe ser extendida a
otros lotes vecinos para confirmar si pueden haber sido afectados.
6.3. Debe ser designada una persona o sector responsable para la
recepción de los reclamos y para la adopción de las medidas
correspondientes.
6.4 Debe existir un procedimiento escrito que describa las acciones a
ser adoptadas en caso de reclamo de posibles desvíos de calidad de un
producto, incluyendo la necesidad de realizar un probable retiro del
mercado.
6.5 Cuando sea necesario, deben ser adoptadas medidas adecuadas de
seguimiento después de la investigación y evaluación del reclamo.
6.6. Los registros de reclamos deben ser regularmente revisados con la
finalidad de detectar cualquier indicio de problemas específicos o
recurrentes y que exijan mayor atención.
7. RETIRO DE PRODUCTOS
7.1 Debe existir un sistema que retire efectivamente del mercado los
productos que presenten desvíos de calidad que puedan ofrecer riesgo al
usuario.
7.2 Debe ser designada una persona responsable para la coordinación del
retiro del producto del mercado. El responsable técnico debe ser
informado sobre las acciones efectuadas, y la Garantía de Calidad debe
acompañar el proceso.
7.3 Deben existir procedimientos escritos, regularmente revisados y
actualizados, para proceder a cualquier actividad de retiro. Los
procedimientos deben contemplar el destino dado a los productos
retirados, la investigación de las causas de devolución y el registro
de todas las acciones tomadas.
7.4 Las Autoridades Sanitarias competentes nacionales y de los países
para los cuales el producto haya sido enviado deben ser inmediatamente
informadas sobre la decisión de recuperación de producto.
7.5 El proceso de retiro debe ser registrado, incluyendo la
conciliación entre las cantidades distribuidas y las cantidades
retiradas del producto en cuestión, con emisión de un informe final.
7.6 Los productos retirados deben ser identificados y almacenados en un
área segregada y seguras hasta que la definición de su destino final.
8. DEVOLUCIÓN
8.1. Debe ser designada una persona o sector responsable para la recepción de las devoluciones.
8.2 Debe existir un procedimiento para la recepción, almacenamiento e investigación de las causas de devoluciones de productos.
8.3 Los productos devueltos deben ser inspeccionados o analizados, o ambos, antes de ser definido su destino final.
8.4 Deben existir registros de los resultados de la inspección y/o del
análisis de los productos devueltos incluyendo sus destinos finales.
8.5 Después de la inspección o análisis o ambos, de los productos
devueltos, deben ser tomadas medidas adecuadas, incluyendo la
posibilidad de retiro del producto.
8.6 En caso que sea necesario, la verificación deberá ser extendida a los lotes vecinos.
9. AUTOINSPECCIÓN /AUDITORÍA INTERNA
9.1 El objetivo de la autoinspección/auditoria interna es evaluar el
cumplimiento de las BPF en todos los aspectos de la fabricación. El
programa de autoinspección/auditoria interna debe ser diseñado de forma
de detectar cualquier desvío en la implementación de las BPF y de
recomendar las acciones correctivas necesarias.
9.2 Deben ser elaborados procedimientos escritos sobre la
autoinspección/auditoria interna. El programa de
autoinspección/auditoria interna debe englobar por lo menos los
siguientes aspectos:
a) personal;
b) instalaciones;
c) mantenimiento de predios y equipos;
d) almacenamiento de materia prima, material de embalaje, producto semielaborado, producto a granel y producto terminado;
e) equipamientos;
f) producción y control en proceso;
g) control de calidad;
h) documentación;
i) sanitización e higiene;
j) programas de validación y revalidación cuando corresponda;
k) calibración de instrumentos y de sistemas de medidas;
l) retiro de producto del mercado;
m) reclamos;
n) gerenciamiento de residuos;
o) resultados de las autoinspecciones/auditorías internas anteriores y cualquier acción correctiva adoptada.
9.3 El equipo de autoinspección/auditoría interna debe estar formado
por profesionales calificados, con conocimiento en BPF. Los miembros
del equipo pueden ser profesionales de la propia empresa,
independientes del área auditada o especialistas externos.
9.4 Las autoinspecciones/auditorías internas deben ser realizadas con una frecuencia de por lo menos una vez al año.
9.5 Una vez finalizada la autoinspección/auditoría interna debe elaborarse un informe que debe contener:
a) los resultados;
b) evaluaciones y conclusiones;
c) las acciones correctivas cuando corresponda;
d) los plazos para adecuación.
9.6. Las acciones correctivas para las no conformidades expresadas en
el informe de autoinspección/auditoría interna deben ser implementadas
y acompañadas conforme al plan de acción.
10. DOCUMENTACIÓN Y REGISTROS
La empresa debe establecer un sistema de documentación de acuerdo con su estructura organizacional y sus productos.
10.1 La documentación constituye parte esencial del sistema de Garantía
de Calidad y, debe estar relacionada con todos los aspectos de las
Buenas Prácticas de Fabricación. Tiene como objetivo definir las
especificaciones de todos los materiales y productos, los
procedimientos de todas las etapas relacionadas con la fabricación y
control de productos, asegurar la uniformidad de interpretación, evitar
confusiones y errores, con la finalidad de garantizar información
necesaria para la liberación o no de lotes de productos según el
cumplimiento de los prerrequisitos de calidad establecidos, asegurando
la existencia de registros que permitan la trazabilidad.
10.2 Los datos deben ser registrados por medios que ofrezcan seguridad
de las informaciones. Todos los datos deben estar disponibles durante
el período de retención establecido en este Reglamento.
10.3 Las alteraciones realizadas deberán ser registradas.
10.4 Toda la documentación relacionada a las Buenas Prácticas de
Fabricación debe ser elaborada, aprobada, actualizada y distribuida de
acuerdo con los procedimientos escritos. Debe estar disponible y ser
archivada de forma segura. El título, la naturaleza y propósito de los
documentos deben ser definidas. La emisión, revisión, sustitución,
retiro y distribución de los documentos deben ser controlados y
registrados de forma segura.
10.5. Los registros corregidos deben posibilitar la identificación del
dato anterior, estar firmados y fechados por el responsable designado.
Ningún documento debe ser modificado sin autorización previa.
10.6 Los documentos y registros deben tener un período de retención
establecido en procedimientos de tal forma que todas las actividades
significativas referentes a fabricación de productos puedan ser
rastreadas.
10.7 Todos los registros de producción y control deben ser retenidos,
por lo menos un (1) año después del vencimiento de la validez del lote
de producto fabricado.
10.7.1 La empresa debe asegurar que los datos permanezcan íntegros y
accesibles durante ese período. Debe haber registro de las alteraciones
realizadas conforme a un procedimiento de control de documentos y
registros.
10.8 La empresa debe mantener registros de uso, limpieza, sanitización
y mantenimiento de los equipamientos conteniendo la fecha, el horario y
responsable por la realización de la tarea. Cuando sea aplicable deben
mantenerse otras informaciones tales como: producto anterior, producto
actual, número de lote del producto procesado, fase del proceso, estado
de aprobación, cuarentena o rechazado.
10.9 Fórmula Patrón
10.9.1. Debe existir una fórmula patrón para cada producto.
10.9.2 La fórmula patrón debe incluir:
a) el nombre del producto y código interno de referencia cuando corresponda;
b) descripción de la forma del producto;
c) lista de todas las materias primas, con las respectivas
especificaciones y cantidades porcentuales, en conformidad con la
fórmula declarada en el registro/notificación;
d) lista completa de todos los materiales de envase y embalaje
requeridos para un tamaño patrón de lote, incluyendo cantidades,
tamaños y tipos, con código o número de referencia relativos a las
especificaciones para cada material de acondicionamiento;
e) los equipamientos de producción a ser utilizados;
f) procedimiento de Fabricación con las instrucciones detalladas;
g) especificaciones de los controles en proceso con sus respectivas metodologías;
h) especificaciones del producto terminado y cuando sea necesario,
deben ser definidas las condiciones especiales de almacenamiento;
i) cualquier precaución especial a ser observada;
10.10 Registros de los lotes de producción
10.10.1 Debe ser mantenido un registro de producción de cada lote
elaborado. El registro se debe basar en la fórmula patrón aprobada
vigente.
10.10.2 Antes del inicio de la producción debe ser verificado que los
equipos y áreas de trabajo estén exentos de productos fabricados
anteriormente, documentos o materiales no requeridos para la producción
planificada y que los equipos estén limpios y adecuados para el uso.
Esta verificación debe ser registrada.
10.10.3 Durante el proceso de producción, todas las etapas que
requieran controles descriptos en el procedimiento de elaboración deben
ser registradas. Las siguientes informaciones deben estar disponibles
para la trazabilidad de la producción:
a) nombre del producto y código interno del producto, cuando corresponda;
b) lote del producto y del granel cuando corresponda;
c) registro de las principales etapas de producción, incluyendo fechas
y horarios de inicio y término cuando es requerido en el proceso de
elaboración;
d) identificación de el/los operador(es) de las diferentes etapas de producción;
e) número de los lotes y la cantidad de cada materia prima y materiales utilizados;
f) cualquier ocurrencia relevante observada en la producción
incluyendo: detalles como la autorización firmada para cada cambio de
la fórmula de fabricación o instrucciones de producción;
g) los principales equipos utilizados;
h) controles en proceso realizados, identificación de la persona que los haya ejecutado y los resultados obtenidos.
10.11. Procedimientos Operativos Estandarizados (POEs) y sus registros
10.11.1 Deben existir procedimientos y registros para:
a) la recepción de materias primas, material de envase y embalaje;
b) la identificación de las materias primas, productos semielaborados,
productos a granel, productos terminados y materiales de envase y
embalaje almacenados en cuarentena, aprobados o rechazados;
c) el muestreo de las materias primas, materiales de envase y embalaje,
productos semi-elaborados, productos a granel, productos terminados;
d) la definición de la codificación del lote específico para materias
primas, materiales de envase y embalaje y productos terminados;
e) los ensayos de control de calidad realizados, describiendo los métodos y los equipos a ser utilizados;
f) la aprobación o rechazo de materiales y productos y definición de la persona o sector responsable;
g) la calificación de Proveedores
h) las actividades de limpieza y sanitización de materiales,
utensilios, equipos y áreas, incluyendo las frecuencias, los métodos y
los materiales de limpieza a ser utilizados;
i) el almacenamiento y expedición de productos;
j) la calibración y mantenimiento de equipos;
k) el control de plagas, contemplando métodos y materiales empleados y desactivación de envases vacíos.
l) el mantenimiento de los equipamientos de prevención y combate para incendios.
m) las medidas de emergencia en caso de derrames de sustancias tóxicas, corrosivas y otro de potencial peligro.
11. PERSONAL
11.1 La empresa debe tener un organigrama actualizado. Las
responsabilidades funcionales deben estar establecidas y documentadas y
ser claramente comprendidas por todos los involucrados.
11.2 El fabricante debe tener un número suficiente de personas entrenadas y calificadas.
11.3 El fabricante debe mediante un programa escrito y definido,
entrenar a las personas involucradas en las áreas de producción, en los
laboratorios de control de calidad, así como a todo el personal cuyas
actividades puedan interferir en la calidad del producto y en la salud
del trabajador.
11.4 Además del entrenamiento básico sobre las BPF, el personal
recientemente contratado debe participar del programa de integración y
recibir entrenamiento apropiado en cuanto a sus atribuciones y ser
entrenado y evaluado continuamente. El programa de entrenamiento debe
ser aprobado, cuando corresponda, por los responsables de la
Producción, de Control de Calidad y de Garantía de Calidad, siendo
mantenidos registros.
11.5. El personal que trabaja en áreas donde son manipulados materiales
tóxicos, corrosivos, cáusticos e inflamables debe recibir entrenamiento
específico.
11.6. Deben existir planificación y cronograma de los entrenamientos
del personal, así como el registro de los entrenamientos realizados.
11.7 El concepto de Garantía de Calidad y todas las medidas capaces de
mejorar su comprensión y su implementación deben ser ampliamente
discutidos durante el entrenamiento.
11.8 Los responsables por la producción y control de calidad deben ser independientes uno del otro.
11.9 La responsabilidad técnica debe ser ejercida por profesional
habilitado. En ausencia del responsable técnico esa función debe ser
ejercida por una persona previamente designada.
11.10 Deberán ser establecidas las responsabilidades funcionales para las siguientes actividades:
a) autorización de los procedimientos y documentos, inclusive sus actualizaciones;
b) seguimiento y control del ambiente de fabricación;
c) higiene;
d) calibración de instrumentos analíticos;
e) entrenamiento, incluyendo la aplicación de los principios de garantía de calidad;
f) aprobación y seguimiento de los proveedores de materiales;
g) aprobación y seguimiento de los fabricantes contratados;
h) especificaciones y seguimiento de las condiciones de almacenamiento de materiales y productos;
i) archivo de documentos/ registros;
j) seguimiento del cumplimento de las BPF;
k) inspección, investigación y muestreo, de modo de hacer un
seguimiento de los factores que puedan afectar la calidad del producto;
l) asegurar que sean realizadas las validaciones cuando sean necesarias.
11.11 Deben también ser establecidas las responsabilidades funcionales para la producción:
a) asegurar que los productos sean producidos y almacenados de acuerdo
con los procedimientos apropiados, con la calidad exigida;
b) aprobar y asegurar la implementación de las instrucciones relativas
a las operaciones de producción, inclusive los controles en proceso;
c) asegurar que los registros de producción sean evaluados y firmados
por el personal designado, antes que sean colocados a disposición de
control de calidad;
d) verificar el mantenimiento de las instalaciones y los equipos;
e) asegurar que las calibraciones y el control de los equipos sean
ejecutados y registrados, y que los informes estén disponibles;
f) asegurar que se haya realizado el entrenamiento inicial y continuo
del personal del área de producción y que el mismo sea adecuado a las
necesidades.
11.12 El responsable por el Control de calidad tendrá las siguientes responsabilidades:
a) aprobar o rechazar las materias primas, los materiales de embalaje, semi-elaborados, a granel y terminados;
b) asegurar que sean realizados todos los ensayos necesarios;
c) aprobar las instrucciones para muestreo, las especificaciones, los
métodos de ensayo de los procedimientos de Control de Calidad;
d) aprobar y hacer un seguimiento de los ensayos realizados por terceros previstos por contrato;
e) hacer un seguimiento del mantenimiento de las instalaciones y de los equipos;
f) asegurar que sean realizadas las calibraciones de los equipos de control;
g) asegurar que sean realizados entrenamientos iniciales y continuos
del personal del área de Control de Calidad, de acuerdo con las
necesidades del sector;
h) mantener registros completos de los ensayos y los resultados de cada
lote de material analizado de forma de emitir un informe analítico
siempre que sea necesario;
i) asegurar la correcta identificación de los reactivos y materiales;
j) certificar la ejecución de la calificación de los equipos del laboratorio, cuando sea necesario.
12. INSTALACIONES
12.1 La empresa debe estar construida en un local compatible con las
actividades desempeñadas y disponer de una planta arquitectónica
aprobada por la autoridad sanitaria competente, con informaciones
necesarias, tales como: área del terreno, área construida, tipo de
construcción e instalaciones destinadas a la fabricación de los
productos.
12.2 Las instalaciones deben ser localizadas, diseñadas, construidas,
adaptadas y mantenidas de forma que sean adecuadas a las operaciones a
ser ejecutadas. Su diseño debe minimizar el riesgo de errores y
posibilitar la limpieza y mantenimiento, de modo de evitar la
contaminación cruzada, o acumulo de polvo y suciedad o cualquier efecto
adverso que pueda afectar la calidad de los productos.
12.3 La limpieza de las áreas y la sanitización, cuando sea necesaria,
deben ser realizada conforme a procedimientos y deben ser mantenidos
los registros correspondientes.
12.4 Las instalaciones deben ser mantenidas en buen estado de conservación, higiene y limpieza.
12.5 Debe asegurarse que las operaciones de mantenimiento y reparación no representen riesgo a la calidad de los productos.
12.6 Los alrededores de los edificios deben estar limpios y en buen estado de conservación.
12.7 El suministro de energía eléctrica, iluminación, aire
acondicionado o ventilación, deben ser apropiados, de modo de no
afectar directa o indirectamente los productos durante los procesos de
fabricación y almacenamiento o el funcionamiento adecuado de los
equipos.
12.8 Las instalaciones deben asegurar la protección contra el ingreso
de insectos y otros animales, manteniendo un programa de prevención y
combate de los mismos, con registros.
12.9 Deben existir instalaciones de seguridad contra incendio, de acuerdo a la legislación específica vigente.
12.10 Los productos raticidas, insecticidas, agentes fumigantes y
materiales sanitizantes deben ser utilizados de manera de no contaminar
equipos, materias primas, materiales de embalaje, materiales en proceso
o productos terminados.
12.11 Los desagües deben ser adecuados, diseñados de manera de prevenir
el reflujo. Siempre que sea posible, los canales abiertos deben ser
evitados. En caso que los canales abiertos sean necesarios deben ser de
fácil limpieza.
12.12 La fabricación de productos domisanitarios con diferentes
aplicaciones solamente puede ser realizada en instalaciones o
equipamiento compartido, siempre que hayan sido hechos,
obligatoriamente, análisis de riesgo y validación de limpieza.
13. SISTEMAS E INSTALACIONES DE AGUA
13.1 La fuente de provisión de agua debe garantizar el abastecimiento con la cantidad y calidad adecuadas.
13.2 La empresa debe definir claramente las especificaciones
físico-químicas y microbiológicas del agua utilizada en la fabricación
de los productos, debiendo cumplir como mínimo los patrones
microbilógicos de potabilidad.
13.2.1 Solamente el agua que esté dentro de las especificaciones
establecidas debe ser utilizada en la fabricación de los productos.
13.3 Las cañerías utilizadas para el transporte de agua deben presentar un buen estado de conservación y limpieza.
13.4 De ser necesario, debe ser realizado tratamiento del agua
previamente al almacenamiento, de forma de atender las especificaciones
establecidas.
13.5 Deben existir procedimientos y registros de operación, limpieza,
higienización, mantenimiento del sistema de tratamiento y distribución
de agua;
13.6 Deben existir procedimientos y registros de seguimiento de la
calidad del agua. El seguimiento debe ser periódico en los puntos
críticos del sistema de agua;
13.7 En caso que sean necesarios patrones de calidad específicos,
definidos de acuerdo con las finalidades de uso de cada producto, el
agua debe ser tratada de forma de cumplirlos.
13.7.1 Deben existir investigaciones, acciones correctivas y
preventivas para resultados del seguimiento del agua fuera de las
especificaciones establecidas. Deben ser mantenidos registros de las
investigaciones y acciones adoptadas.
13.8 La circulación de agua debe ser efectuada por cañerías u otro
medio que ofrezca seguridad en cuanto a mantenimiento de los patrones
establecidos de calidad del agua.
13.9 En caso de almacenamiento de agua deben existir dispositivos o tratamientos que eviten la contaminación microbiológica.
14. ÁREAS AUXILIARES
14.1 Las salas de descanso, comedor, vestuarios, sanitarios y áreas de
mantenimiento deben estar separadas de las áreas de producción.
14.2 Los vestuarios, lavatorios y los sanitarios deben ser de fácil
acceso y en cantidad suficiente para el número de usuarios, en
condiciones de higiene apropiada, provistos con jabón y toallas o
secadores. Los sanitarios no deben tener comunicación directa con las
áreas de producción y almacenamiento.
14.3 Las áreas de mantenimiento deben estar situadas en locales
separados de las áreas de producción. Si las herramientas y piezas de
reposición son mantenidas en las áreas de producción, las mismas deben
estar en salas o armarios o espacios reservados para ese fin.
14.4 Las cañerías de agua, vapor, gas, aire comprimido y electricidad
deben estar identificados conforme a la legislación vigente.
15. RECEPCIÓN Y ALMACENAMIENTO
15.1 La adquisición de materiales debe ser planeada y controlada para
que atienda las necesidades de la calidad. Los requisitos deben estar
claramente establecidos y documentados, informados y comprendidos por
los proveedores.
15.2 Las áreas de depósito deben tener capacidad suficiente para
posibilitar el almacenamiento ordenado de varias categorías de
materiales y productos: materias primas, materiales de embalaje,
productos intermedios, productos a granel y productos terminados, en su
condición de cuarentena, aprobado, rechazado, devuelto o retirado.
15.3 Las áreas de depósito deben asegurar las condiciones de
almacenamiento exigidas para los materiales y productos. Deben ser
limpias, secas y mantenidas a temperaturas compatibles con los
materiales almacenados. Cuando fueran exigidas condiciones especiales
de almacenamiento, temperatura y humedad, tales condiciones deben ser
provistas, verificadas, monitoreadas y registradas.
15.4 Los pisos, paredes y techos deben ser de fácil limpieza, material resistente y deben estar en buen estado de conservación.
15.5 Las instalaciones de los depósitos deben estar protegidas contra
la entrada de roedores, insectos, aves y otros animales, debiendo
existir un sistema para combatir a los mismos.
15.6 En caso de desvíos en relación a los parámetros establecidos debe
ser hecha investigación para determinar las causas, debiendo ser
tomadas acciones preventivas y correctivas en relación a las causas
identificadas, procediéndose a registrar las mismas.
15.7 Todas las actividades ejecutadas en las áreas de almacenamiento
deben cumplir con los procedimientos previamente definidos, con
registro de las operaciones críticas.
15.8 Las balanzas deben ser calibradas y verificadas conforme a la
periodicidad establecida y deben ser mantenidos los registros de esas
actividades.
15.9 Debe existir un área, y/o sistema, que delimite o restrinja el uso
de los materiales y productos respetándose el “status” previamente
definido.
15.10 Los materiales y productos rechazados, retirados y devueltos
deben estar identificados como tal y almacenados separadamente en área
restringida o segregada. Cualquier otro sistema que sustituya la
identificación a través de etiquetas o la segregación debe ofrecer
seguridad.
15.11 El sistema de registro y control de almacenamiento de los
productos intermediarios y a granel debe incluir el tiempo máximo de
almacenamiento permitido antes de su embalaje.
15.12 El sistema de registro y control de expedición debe observar la
correspondiente relación secuencial de lotes y plazo de validez cuando
corresponda.
15.13 Los materiales que presentan riesgos de incendio o explosión y
otras sustancias peligrosas deben ser almacenados en áreas seguras y
protegidas, debidamente segregados e identificados, de acuerdo con la
legislación específica vigente.
15.14. Los materiales deben ser almacenados bajo condiciones y períodos
adecuados de modo de preservar su integridad e identidad. El
almacenamiento debe ser controlado para que la rotación obedezca la
regla: lo primero que expira es lo primero que sale (PEPS), cuando
corresponda.
15.15 Debe existir un sistema para el control del stock. En caso sean
utilizados sistemas informatizados para gerenciamiento de materiales y
productos, la empresa debe comprobar la seguridad del sistema.
15.16 La empresa debe realizar inventarios periódicos o sistema similar, manteniendo registros de los mismos.
15.17 Los materiales y productos almacenados deben estar aislados del
piso y separados de las paredes, para facilitar la limpieza y
conservación.
15.18 Los materiales y productos deben estar identificados
correctamente por su fabricante/proveedor. El rótulo o etiqueta de
identificación debe estar debidamente adherido al cuerpo del recipiente
que lo contiene.
15.19 Cuando se realiza la recepción, cada lote de materiales y
productos debe estar identificados hasta el final de su utilización.
15.20 Los materiales y productos deben permanecer en cuarentena
debidamente identificados como tal, antes de su liberación por Control
de Calidad. En los casos de almacenamiento controlado por sistema
informatizado, su uso debe ser bloqueado hasta ser liberados por la
persona autorizada.
15.21 Los rótulos, etiquetas o controles por sistema electrónico de los
materiales y productos deben permitir su identificación correcta y
visualización de su estado.
15.22 Las etiquetas o sistemas de identificación deben disponer las siguientes informaciones:
a) nombre del material o producto y el respectivo código interno de referencia, cuando corresponda;
b) número de lote asignado por el proveedor o, cuando sea aplicable, el número dado por la empresa cuando lo recepciona;
c) situación de los materiales: cuarentena, en análisis, aprobado, rechazado o devuelto;
d) fecha de validez;
e) nombre del proveedor.
15.23 Solamente las materias primas liberadas por Control de Calidad pueden ser usadas para la fabricación de productos.
15.24 Debe ser respetado el plazo de validez establecido por el
fabricante de las materias primas. El reanálisis de las materias primas
en almacenamiento sirve solamente para la confirmación del
mantenimiento de sus especificaciones y no puede ser usada para
extender el plazo de validez.
15.25 El almacenamiento debe ser realizado con el debido orden y
seguridad, evitando posibles mezclas en su control y expedición, así
como accidentes en su manipulación.
15.26 Los productos deben estar apilados con seguridad.
15.27 La empresa debe poseer procedimientos/sistema para asegurar que
los materiales y productos terminados no sean utilizados con su plazo
de validez expirado.
15.28 La empresa debe poseer procedimientos de verificación e
inspección de los materiales y productos de forma de garantizar la
recepción de materiales y productos dentro de los requerimientos
definidos.
15.29 Si un único envío de materiales y productos contiene lotes
distintos, cada lote debe ser considerado separadamente para el
muestreo y ensayos de liberación.
15.30 Las materias primas deben ser recibidas con los respectivos certificados de análisis del fabricante/proveedor.
15.31 En las áreas de recepción y expedición los materiales deben ser
protegidos de las variaciones climáticas que pongan en riesgo la
integridad de los materiales manipulados.
15.32 Las áreas de recepción deben ser adecuadas, equipadas de forma de
permitir que los recipientes de materiales recibidos estén limpios
externamente antes de ser almacenados.
16. MUESTREO DE MATERIALES
16.1 El muestreo debe ser realizado en área definida, por personas
autorizadas, de modo de evitar cualquier tipo de contaminación
microbiológica o cruzada.
16.2. Las muestras deben ser representativas del lote de material
recibido, en caso de recibir más de un lote del mismo material, los
mismos deben ser muestreados separadamente.
16.3. El número de los recipientes muestreados y el tamaño de la muestra deben estar basados en un plan de muestreo.
16.4 El muestreo debe ser conducido obedeciendo los procedimientos
aprobados de forma de garantizar la protección de las muestras de
contaminaciones.
16.5. Todos los equipos (instrumentos, recipientes, utensilios)
utilizados en los procesos de muestreo que entraran en contacto con los
materiales deben estar limpios, sanitizados cuando sea aplicable y
guardados en lugares apropiados, debidamente identificados.
16.6 Las etiquetas o sistema de identificación deben proporcionar las siguientes informaciones:
a) nombre y/o código interno del material muestreado;
b) número de lote;
c) identificación de la persona que tomó la muestra;
d) fecha en que la muestra fue tomada.
16.7 Los recipientes de los cuales fueron retiradas las muestras deben ser identificados.
16.8 Los Procedimientos Operativos Estandarizados relativos a muestreo deben incluir, como mínimo:
a) identificación de la función/cargo de la persona designada para tomar la muestra;
b) método o criterio de muestreo:
i. Número de recipientes;
ii. Cantidad de material;
iii. . Instrumentos utilizados para el muestreo;
c) equipamiento a ser usado para el muestreo y EPI (equipamiento protección individual), cuando sea necesario;
d) tipo de embalaje para la muestra, condición de muestreo (sea aséptico o no) y rótulo;
e) cualquier precaución especial a ser observada;
f) Instrucciones para limpieza y almacenamiento de los equipos de muestreo;
g) condición de almacenamiento de las muestras;
h) destino del sobrante del muestreo;
i) condiciones ambientales del lugar de muestreo (luz, temperatura y humedad) cuando corresponda;
j) período de retención de las muestras;
k) identificación de la muestra;
l) instrucciones para cualquier subdivisión de la muestra que fuera necesaria.
17. PRODUCCIÓN/ELABORACIÓN
17.1 La empresa debe establecer procedimientos para la seguridad de las instalaciones en las áreas de producción.
17.2 Las condiciones externas y las áreas destinadas a producción de
domisanitarios deben permitir la adecuada limpieza y mantenimiento.
17.3 Las áreas de producción deben estar provistas de la
infraestructura necesaria, lo que incluye espacio, instalaciones,
equipos, materiales adecuados, personal calificado y debidamente
entrenado para ejecución de las actividades, procedimientos
operacionales e instrucciones de trabajo aprobadas, además de personal
calificado y equipos adecuados para la realización de controles en
proceso.
17.4 La distribución de las áreas de producción debe ser ordenada y
racional. Las instalaciones físicas deben estar dispuestas, según un
flujo operacional continuo, con distribución adecuada para evitar
mezcla o contaminación cruzada.
17.5 Las áreas productivas deben ser de tamaño compatible con el
volumen de operaciones realizadas y con las identificaciones
necesarias. En los casos de productos que por sus características
puedan provocar contaminación cruzada deben existir áreas separadas
para elaborar y envasar. Toda el área de circulación debe estar libre
de obstáculos.
17.6 Los sectores deben ser distribuidos de modo de permitir que la
producción ocurra de forma adecuada, evitando mezclas o contaminación
cruzada.
17.7 Las cañerías, luminarias, puntos de ventilación y otras
instalaciones, deben ser adecuadas de modo de facilitar la limpieza y
mantenimiento. Siempre que sea posible el acceso para mantenimiento
debe estar localizado externamente a las áreas de producción. Cuando no
fuera posible el acceso externo para los servicios, los procedimientos
de mantenimiento deben ser ejecutados de forma de minimizar el riesgo
de contaminación y/o que comprometa la calidad del producto.
17.8 La iluminación y ventilación deben ser suficientes y adecuadas
para la ejecución de los procesos productivos y deben estar de acuerdo
con la legislación vigente.
17.9 La temperatura y humedad deben ser monitoreadas, registradas y
controladas, cuando sea necesario y deben ser compatibles las
condiciones de estabilidad de los materiales y de los productos
terminados.
17.10. Cuando sea necesario las áreas deben poseer sistemas de
extracción de aire adecuados y que garanticen la protección contra la
contaminación cruzada.
17.11 La empresa debe disponer de procedimientos para la limpieza e
higienización de las áreas de producción, de los equipos y sus
registros. Debe existir un lugar separado para guardar los materiales
utilizados en la limpieza y mantenimiento.
17.12 Los desagües deben ser sifonados, desinfectados frecuentemente y
mantenidos cerrados. Deben ser lisos para facilitar la limpieza y la
desinfección.
17.13 Los basureros deben ser identificados, cerrados y vaciados de acuerdo con la necesidad.
17.14. En las áreas productivas deben estar disponibles equipos de protección individual y colectivo (EPI / EPC).
17.15. Antes de iniciar un proceso de producción, debe ser verificado
si los equipos y el lugar de trabajo están libres de productos
anteriormente producidos, así como los documentos y materiales
necesarios para el proceso planeado. Además de eso, debe ser verificado
si los equipos están limpios y adecuados para el uso. Las
verificaciones de esos ítems deben ser registradas.
17.16 Las ventanas u otras aberturas de las áreas de producción o
envasado deben estar protegidas de modo de evitar posibilidad de
contaminación.
17.17. Áreas de pesada y medidas
17.17.1 La empresa, cuando corresponda, debe poseer un área definida
para las actividades de pesada y medidas de materias primas destinadas
a la producción. Cuando haya riesgo para los trabajadores o de
contaminación cruzada el área debe ser separada físicamente de las
demás dependencias.
17.17.1.1 Las áreas destinadas a medidas, cuando corresponda y la
pesada de las materias primas pueden estar localizadas en el depósito o
en el área de producción, debiendo las mismas ser diseñadas y separadas
para ese fin, poseyendo un sistema de extracción de aire independiente
y adecuado, que evite la ocurrencia de contaminación cruzada y
ambiental.
17.17.2 Las balanzas y recipientes de medidas deben ser calibrados
regularmente, de acuerdo con un programa de calibración preestablecido
y presentar registros de las calibraciones. Debe ser establecida la
periodicidad de las verificaciones.
17.17.3 Las áreas de pesada y medidas deben estar constantemente limpias.
17.17.4 Las operaciones de pesada deben ocurrir de acuerdo con la orden de producción, según procedimiento específico.
17.17.5 Los recipientes o embalajes externos de las materias primas a
ser pesadas y/o medidas deben estar limpios antes de entrar en las
áreas de pesada. Después de la pesada o medida, esos recipientes deben
ser mantenidos cerrados.
17.17.6 A fin de evitar mezclas, los materiales pesados y/o medidos
deben ser inmediatamente identificados por medio de etiquetas o
sistemas de identificación conteniendo el nombre, lote de la materia
prima y la cantidad pesada o medida, pudiendo agregar el código interno.
17.17.7 Los materiales medidos o pesados deben ser segregados físicamente por lote u orden de producción.
17.17.8. Los utensilios de pesada y medidas deben estar limpios,
identificados en cuanto a su estado de limpieza y guardados en lugar
que asegure su integridad.
17.17.9 Todas las actividades de pesada, verificación, calibración y mantenimiento deben ser registradas.
17.17.10 El recipiente de materia prima que haya sido pesado y que por
no ser utilizado retornará al depósito debe ser cerrado e identificado
adecuadamente.
17.18 Equipamientos
17.18.1 Las balanzas e instrumentos de medida de las áreas de
producción y de control de calidad deben tener la capacidad y la
precisión requerida.
17.18.2 Las balanzas y demás equipos de precisión y medida utilizados
en el área de producción deben estar calibrados y verificados antes de
su uso. Deben ser realizadas calibraciones periódicas, de acuerdo con
un programa de calibración preestablecido.
17.18.3 Los equipos deben ser diseñados, construidos, adaptados,
instalados, localizados y mantenidos de forma de facilitar las
operaciones a ser realizadas. Los equipos no deben presentar riesgos
para la calidad de los productos. Las partes que entran en contacto con
el producto no deben ser reactivas, aditivas o absorbentes de forma de
no interferir en la calidad del producto.
17.18.4 El proyecto y la localización de los equipos deben minimizar
los riesgos de errores y permitir la limpieza y mantenimiento adecuados
de manera de evitar la contaminación cruzada, acumulo de polvo y
suciedad, y en general, evitar todo efecto que pueda influir
negativamente en la calidad y seguridad de los productos.
17.18.5 Las áreas de circulación entre los equipos deben ser mantenidas
libres. Los procesos de limpieza y lavado de los equipos no deben
constituir fuente de contaminación del producto y deben ser registrados.
17.18.6. Todo equipo en desuso o con defectos debe ser retirado de las
áreas de producción, caso contrario, debe estar debidamente
identificado.
17.18.7 Todos los equipos deben estar debidamente identificados y sometidos a limpieza y sanitización, conforme a procedimiento.
17.18.8 La empresa debe establecer un programa de mantenimiento
preventivo de equipos. Las actividades de mantenimiento deben ser
registradas.
17.18.9 Las cañerías fijas deben estar claramente identificadas en
cuanto a su contenido y donde sea aplicable, la dirección del flujo.
17.18.10 Las cañerías, conexiones, dispositivos o adaptadores para
gases o líquidos peligrosos deben estar identificados y no deben ser
intercambiables.
17.19 Áreas de Elaboración/Procesos
17.19.1 Los procesos productivos deben ser ejecutados a partir de un
planeamiento de producción. Todos los lotes producidos deben seguir una
orden de producción y esta debe coincidir con la fórmula patrón del
producto.
17.19.2 Es recomendable que la indumentaria utilizada en el área de
producción sea de uso exclusivo de este sector, no siendo recomendable
la circulación por otras dependencias de la fábrica con esta
indumentaria.
17.19.3 Antes de iniciar cualquier operación de producción, se debe asegurar que:
a) toda documentación pertinente esté disponible;
b) todas las materias primas estén disponibles y aprobadas;
c) los equipamientos deben estar disponibles y en condiciones operacionales;
d) los equipos utilizados en la producción deben estar debidamente
identificados con nombre y/o codificación y lote del producto que está
siendo fabricado;
e) en caso de procesos continuos o dedicados la identificación con
nombre y/o codificación y lote del producto puede estar disponible en
los registros de fabricación;
f) el área de producción debe estar liberada de acuerdo a un
procedimiento establecido de forma de evitar mezclas con materiales de
operaciones anteriores.
17.19.4 El número de lote debe ser atribuido para cada partida de
producción a granel. Este no precisa ser necesariamente el número que
se incluye en el rótulo del producto acabado, siempre que se defina
claramente la vinculación entre ambos.
17.19.5 La tercerización de etapa(s) productiva(s) o de control de
calidad debe(n) ser registrada(s) y estar de acuerdo con la legislación
vigente.
17.19.6 Todas las etapas de producción deben ser registradas por el
operador, en el momento de realización de la actividad, y las etapas
críticas deben ser monitoreadas o verificadas de acuerdo con el
procedimiento establecido.
17.19.7 La identificación de los productos a granel debe incluir:
a) el nombre y/o código de identificación;
b) el número de partida o lote;
c) las condiciones de almacenamiento cuando fueran críticas para asegurar la calidad del producto.
17.19.8 Todos los controles de procesos y los correspondientes límites
de aceptación deben estar definidos. Los mismos deben ser ejecutados de
acuerdo con lo establecido en procedimientos escritos. Cada resultado
que esté fuera del límite según el criterio de aceptación debe ser
informado, registrado e investigado.
17.19.9 El reprocesamiento de productos solamente puede ser permitido
si la calidad del producto terminado no fuera afectada, si las
especificaciones son cumplidas y si la operación fue realizada de
acuerdo con los procedimientos autorizados y definidos después de la
evaluación de los riesgos involucrados. Debe ser mantenido un registro
de reprocesamiento. Cualquier lote reprocesado debe recibir
identificación que permita su trazabilidad.
17.19.10 Cuando el proceso no fuera continuo, debe haber un área
delimitada o separada para el almacenamiento de productos
semi-elaborados o a granel, de acuerdo con las especificaciones del
producto y procedimiento que defina el tiempo máximo de almacenamiento.
17.19.11 Debe ser efectuada la limpieza de los equipos después de cada
producto fabricado. La elaboración en campaña sin la limpieza de los
equipos solamente podrá ser realizada de acuerdo con el procedimiento
descripto que determine los controles en proceso lote a lote y el
número máximo de lotes secuenciales permitidos.
17.20 Áreas de envasado/rotulado
17.20.1 Debe existir un área apropiada o lugar específico para el
envasado/embalaje de productos. La distribución de los equipos debe ser
ordenada y racional.
17.20.2 Las instalaciones físicas para el envasado/embalaje de los
productos deben ser diseñadas de forma de evitar la ocurrencia de
mezclas entre diferentes productos y lotes.
17.20.3 Antes del inicio de operaciones de embalaje se debe asegurar
que el área de trabajo, las líneas de embalaje, impresoras y equipos
estén limpios y exentos de productos, materiales o documentos de
operaciones anteriores. La liberación del área debe ser realizada de
acuerdo con procedimiento escrito y una lista de verificación con
registros.
17.20.4 Los rótulos deben ser inspeccionados antes de ser entregados a
la línea de embalaje. En el proceso de rotulado se debe verificar si
los rótulos corresponden al producto, así como el número de lote y la
fecha de vencimiento del producto.
17.20.5 El producto a granel debe mantenerse cerrado durante el proceso
de envasado, siendo abierto solamente cuando sea necesario. Debe
existir identificación del producto (nombre y/o codificación y lote) de
forma visible en los equipos y de cada línea de envasado.
17.20.6 Es recomendable la verificación de la relación entre el
rendimiento teórico y el real y si hubiera discrepancia con los
parámetros establecidos, justificar por escrito.
17.20.7 Cuando sea aplicable y en conformidad con procedimiento
interno, los productos después del envasado/embalaje deben quedar en
cuarentena, debidamente identificados en cuanto a su estado hasta la
liberación por Control de Calidad/Garantía de Calidad.
17.20.8 El material codificado remanente del envasado/embalaje debe ser destruido y se debe registrar la operación.
17.20.9 Todos los controles de procesos y sus correspondientes límites
de aceptación deben estar definidos. Los mismos deben ser ejecutados de
acuerdo con lo establecido en procedimientos escritos. Cada resultado
que este fuera del límite según el criterio de aceptación debe ser
informado, registrado e investigado.
17.20.10 Todos los materiales de envase y embalaje que no hayan sido
utilizados y que sean reenviados al depósito deben estar identificados
y registrados.
17.20.11 En los casos en que el envasado y rotulado no sean continuos,
deben ser adoptadas medidas de identificación y segregación para evitar
mezclas o errores de rotulado.
17.21 Gestión de Residuos
17.21.1 Deben existir procedimientos escritos de gestión de residuos de
acuerdo a la legislación vigente, los cuales deben ser de conocimiento
previo de los responsables involucrados.
17.21.2 Los efluentes y residuos resultantes de la fabricación, de los
edificios y de las áreas circundantes deben estar dispuestos de manera
segura y sanitaria hasta su destino. Los recipientes y las cañerías
para el material de descarte deben estar identificados.
17.21.3 Los efluentes y residuos deben ser identificados y clasificados
según su naturaleza. Deben establecerse los destinos, los controles
efectuados y el lugar de vertido de los residuos y efluentes tratados.
Deben registrarse los controles realizados y su frecuencia.
17.21.4 El manejo y la disposición de residuos no deben impactar las operaciones de producción o la calidad de los productos.
18. CONTROL DE CALIDAD
18.1 La empresa debe poseer laboratorio de Control de Calidad, propio e
independiente de la producción. Para los casos de tercerización de
ensayos de Control de Calidad la empresa debe seguir la legislación
vigente.
18.2 Los requisitos mínimos para el Control de Calidad son los siguientes:
a) los ensayos deben ser ejecutados de acuerdo con procedimientos;
b) los instrumentos de precisión deben ser calibrados a intervalos definidos utilizando un patrón de referencia certificado;
c) poseer equipos adecuados para los procedimientos de ensayos
previstos y en número suficiente para el volumen de las operaciones;
d) personal calificado y entrenado;
e) deben ser registrados los resultados de los ensayos de control de
materias primas, materiales de embalaje y productos terminados.
18.3 Las responsabilidades principales de Control de Calidad no deben
ser delegadas. Estas responsabilidades deben ser definidas y
documentadas contemplando como mínimo las siguientes actividades:
a) participar de elaboración, actualización y/o revisión de:
i. especificaciones y métodos analíticos para materias primas, materiales de empaque, controles en proceso, producto terminado;
ii. procedimientos de muestreo;
iii. procedimiento para el monitoreo ambiental de áreas productivas;
iv. procedimiento para evaluar y almacenar los patrones de referencia.
b) aprobar o rechazar materias primas, materiales de empaque, productos semi-elaborados, producto a granel y producto terminado;
c) mantener registros completos de ensayos y resultados de cada lote de
material analizado de manera de emitir un informe de análisis, siempre
que sea necesario;
d) asegurar la ejecución de todos los ensayos necesarios;
e) participar en la investigación de reclamos y devoluciones de productos terminados;
f) asegurar la correcta identificación de reactivos y materiales;
g) participar de la investigación de los resultados fuera de especificación;
h) verificar y registrar el mantenimiento de las instalaciones, así
como la calibración y mantenimiento de los equipos del laboratorio;
i) certificar la ejecución de la calificación de los equipos de laboratorio cuando sea necesario;
j) garantizar la trazabilidad de los procesos realizados bajo su responsabilidad;
k) coordinar los entrenamientos iniciales y continuos de los empleados.
18.4 Los laboratorios de Control de Calidad deben ser separados de las
áreas de producción. Las áreas donde fueran realizados los ensayos
microbiológicos deben contar con instalaciones independientes.
18.5 Los laboratorios de Control de Calidad deben disponer de espacio
suficiente, áreas apropiadas y ser diseñados de acuerdo con la lógica
de las operaciones en ellos realizadas.
18.6 El laboratorio debe ser diseñado considerando la utilización de
materiales de construcción adecuada y la actividad que será
desarrollada, y debe poseer un conjunto de dispositivos que aseguren
las condiciones ambientales para la realización de los ensayos y la
protección de la salud de las personas.
18.7 De ser necesario, deben utilizarse salas y equipos separados para
proteger determinados instrumentos de interferencias eléctricas,
vibraciones, contacto excesivo con humedad y otros factores externos.
18.8 Los procedimientos de los ensayos deben ser aprobados por Garantía
de Calidad y estar disponibles en las unidades responsables para la
ejecución de los mismos.
18.9 Las especificaciones deben ser establecidas por la empresa y estar
debidamente autorizadas y fechadas en relación a los ensayos de las
materias primas, incluyendo agua, materiales de embalaje y productos
terminados. Además, deben ser realizados ensayos en los productos
semi-elaborados y en los productos a granel, cuando sea necesario.
18.10 Deben realizarse revisiones periódicas de las especificaciones.
18.11 La literatura, los manuales de los equipos, los patrones de
referencia y otros materiales necesarios deben estar a disposición del
laboratorio de Control de Calidad.
18.12 El control de Calidad debe tener fácilmente disponible en el sector:
a) especificaciones;
b) procedimientos de muestreo;
c) métodos de ensayo y registros (sean hojas analíticas y/o cuaderno de anotaciones y/o medio electrónico);
d) boletines y/o certificados analíticos;
e) registros de monitoreo ambiental, donde sea especificado;
18.13 Los registros de ensayo deben incluir, por lo menos, los siguientes datos:
a) el nombre y/o codificación del material o producto y cuando sea aplicable, la forma de presentación;
b) lote y nombre del fabricante y/o proveedor;
c) referencias para procedimientos de análisis;
d) resultados analíticos, incluyendo cálculos, observaciones (si son necesarias) y los limites de especificaciones;
e) fecha de los ensayos;
f) identificación de los responsables de la ejecución de los ensayos;
g) fecha e identificación de los responsables de la verificación de los análisis y los cálculos cuando sea aplicable;
h) resultado de aprobación o rechazo del material o producto, firmado por persona autorizada.
18.14 En el informe de análisis deben constar como mínimo:
a) nombre y/o codificación de la materia prima o del producto;
b) lote;
c) fecha de fabricación;
d) fecha de vencimiento cuando corresponda;
e) cada ensayo ejecutado, incluyendo los limites de aceptación, los
resultados obtenidos y cuando corresponda, referencias de la
metodología analítica utilizada;
f) fecha de emisión del informe, identificación y firma de la persona autorizada;
g) identificación del fabricante, cuando corresponda.
18.15 El Control de calidad/Garantía de Calidad es responsable de
asegurar que sean ejecutados los controles necesarios para el muestreo
y ensayo, para que todos los materiales y productos terminados sean
liberados solamente si cumplen todos los requisitos de los criterios de
aceptación especificados. Estos controles incluyen revisión de la
documentación del lote, muestras de retención, evaluación y
almacenamiento de patrones de referencia, revisión de especificaciones
de materiales y productos, pudiendo también incluir el monitoreo
ambiental.
18.16 El laboratorio de control de calidad debe realizar todos los
ensayos necesarios para confirmar que las materias primas, materiales
de envase y embalaje, productos a granel, semielaborados y terminados
cumplan con los criterios de aceptación establecidos. Se admite
prescindir de la realización de ensayos cuando el proveedor fuera
calificado.
18.17 El Control de Calidad debe establecer las especificaciones para
selección y compra de materiales e insumos de uso del laboratorio.
18.18 Los reactivos y las soluciones volumétricas adquiridas y/o
preparadas deben estar identificadas y de acuerdo con las
especificaciones. Los procedimientos de preparación de los mismos deben
definir su validez de uso.
18.19 En caso de ensayos de pureza e identificación de una sustancia
química, el patrón de referencia certificado debe estar disponible.
18.20 Los patrones de referencia internos deben ser verificados
periódicamente en cuanto al mantenimiento de sus propiedades, debiendo
mantenerse los respectivos registros.
18.21 Las sustancias químicas de referencia deben ser apropiadas para
la realización de los ensayos de los productos terminados, con origen
documentado y las mismas mantenidas en las condiciones de
almacenamiento recomendadas por el fabricante.
18.22 Cuando una sustancia química de referencia no estuviera
disponible, otro patrón debe ser establecido. Deben realizarse ensayos
de identificación y pureza para este patrón. Debe mantenerse la
documentación de los resultados de los ensayos.
18.23 Los reactivos deben ser debidamente identificadas debiendo
contener en su rótulo como mínimo las siguientes informaciones: nombre,
concentración, fecha de validez y/o períodos de almacenamiento
recomendados, fecha de preparación, identificación del técnico
responsable de la preparación y cuando corresponda, el factor de
corrección.
18.24 Todos los resultados de los controles deben revisarse y decidida
la situación del material en cuanto a aprobación, rechazo o pendiente.
18.25 Especificaciones para materiales y productos
18.25.1 Todos los ensayos deben seguir las instrucciones establecidas
por los procedimientos escritos y aprobados para cada material o
producto.
18.25.2 Las especificaciones de las materias primas, de los materiales
de envase primario y de los materiales impresos, deben poseer una
descripción, incluyendo, como mínimo:
a) denominación y/o nombre químico de la materia prima;
b) nombre y/o código interno de referencia;
c) referencia de las literaturas reconocidas, cuando corresponda;
d) requisitos cuantitativos y cualitativos con los respectivos limites de aceptación;
e) modelo del material impreso, cuando corresponda.
18.25.3 Los materiales de embalaje deben cumplir con las
especificaciones. El material debe ser examinado en relación a defectos
físicos visibles y críticos, atendiendo las especificaciones requeridas.
18.25.4 Las especificaciones de los productos intermedios y de los
productos a granel deben estar disponibles siempre que estos materiales
fueran adquiridos o expedidos, o si los datos sobre los productos
intermedios tuvieran que ser utilizados en la evaluación del producto
final.
18.25.5 Deben establecerse especificaciones para productos terminados
de acuerdo con criterios de aceptación y estas deben ser consistentes
con el proceso de fabricación.
18.25.6 Cuando un producto terminado tenga una especificación
microbiológica, los límites de aceptación de recuento total de
microorganismos y microorganismos patógenos deben estar en conformidad
con la legislación vigente.
18.26 Análisis de Materiales y Productos
18.26.1 Antes que los materiales y productos sean liberados para el
uso, Control de Calidad debe garantizar que los mismos sean analizados
en cuanto a la conformidad con las especificaciones.
18.26.2 Deben utilizarse solamente las materias primas liberadas por
Control de Calidad y que estén dentro de los respectivos plazos de
validez.
18.26.3 Los productos que no cumplan con las especificaciones
establecidas deben ser rechazados. Si es viable, pueden ser
reprocesados, debiendo ser previamente autorizados y realizados de
acuerdo con procedimientos definidos. Los productos reprocesados deben
cumplir con todas las especificaciones y criterios de calidad antes de
ser aprobados y liberados.
18.26.4 Deben existir equipos de seguridad disponibles, los cuales deben ser verificados/probados regularmente.
18.27 Laboratorio Microbiológico
18.27.1 Control de Calidad del titular del producto debe ser el
responsable por aprobar o rechazar análisis que estén sub contratados
con terceros.
18.27.2 Cuando corresponda, deben realizarse ensayos microbiológicos en
cada lote de producto terminado, respetando los límites de aceptación
presentes en la legislación vigente.
18.27.3 Debe existir un programa de limpieza definido y registrado para
el laboratorio microbiológico, considerando el resultado del monitoreo
ambiental y la posibilidad de contaminación.
18.27.4 La empresa debe garantizar la seguridad de la manipulación y el
descarte de materiales de riesgo biológico y mantener procedimientos y
registros adecuados.
18.27.5 Los procesos de descontaminación y esterilización deben ser
controlados y documentados de forma de garantizar la seguridad y
eficacia de los diferentes procesos.
18.27.6 Los autoclaves deben ser calificados. Para cada ciclo
operacional y cada tipo de carga usado en el/los autoclave(s) deben
realizarse estudios de calificación de performance, manteniéndose los
registros.
18.27.7 Los medios de cultivo deben ser preparados e identificados
según procedimientos escritos debidamente aprobados, teniendo como
referencia las recomendaciones del fabricante.
18.27.8 Los medios de cultivo deben ser analizados en cuanto a la viabilidad de crecimiento en las condiciones requeridas.
18.27.9 Los reactivos (incluyendo los remanentes), medios de cultivo,
diluyentes, entre otros, deben estar identificados. Para permitir la
trazabilidad de estos materiales deben estar disponibles las siguientes
informaciones: nombre, concentración (cuando corresponda), fecha de
validez y/o período de almacenamiento recomendado, fecha de preparación
y responsable por la preparación.
18.27.10 Los cultivos de referencia deben ser adquiridos de fuentes
reconocidas, con presentación de los respectivos certificados.
18.27.11 Deben existir procedimientos escritos para la preparación y
conservación de sub-cultivos para uso como estándares de referencia,
siendo realizados ensayos de identificación y caracterización de las
cepas y de los sub-cultivos.
18.27.12 La toma y manipulación de muestras deben realizarse de acuerdo
con procedimientos escritos de forma de evitar la contaminación del
material.
19. CONTRAMUESTRAS
19.1 Las muestras de productos terminados deben ser retenidas en los
envases originales o en un envase equivalente al material de
comercialización y almacenadas en las condiciones especificadas, en
cantidad suficiente para permitir como mínimo dos análisis completos.
19.2 Las contramuestras deben poseer rótulo conteniendo identificación, lote y fecha de validez.
19.3 Tiempo mínimo de almacenamiento de las contramuestras:
a) Las muestras de materias primas, cuando corresponda, deben ser retenidas hasta el vencimiento de su plazo de validez;
b) Las muestras de productos terminados deben retenerse hasta el vencimiento de su plazo de validez.
MERCOSUR/GMC/RES. Nº 11/15
FARMACOPEA MERCOSUR: ESPECTROFOTOMETRÍA INFRARROJO
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto y las Resoluciones N° 31/11 y 22/14 del Grupo Mercado Común.
CONSIDERANDO:
Que la Farmacopea MERCOSUR tiene como objetivo establecer los
requisitos mínimos de calidad y seguridad de los insumos para la salud,
especialmente de los medicamentos, apoyando las acciones de
reglamentación sanitaria y promoviendo el desarrollo técnico,
científico y tecnológico regional.
Que las especificaciones farmacopeicas establecen, por medio de
monografías, requisitos mínimos para el control de seguridad y calidad
de los insumos, especialidades farmacéuticas, plantas medicinales y
derivados producidos o utilizados en los Estados Partes.
Que las especificaciones farmacopeicas son utilizadas como parámetro
para las acciones de vigilancia sanitaria, incluyendo el registro de
medicamentos, inspecciones y análisis de laboratorio.
Que la Farmacopea MERCOSUR y la producción de patrones propios de
calidad favorecen al desarrollo científico y tecnológico de los Estados
Partes, contribuyendo a la disminución de la dependencia de proveedores
extranjeros y promoviendo a la industria regional.
Que la Farmacopea MERCOSUR debe ser primordialmente sanitaria, con
énfasis en la salud pública, y presentar una metodología analítica
accesible a los Estados Partes, buscando su reconocimiento y
respetabilidad internacional.
Que el diálogo regulatorio y la integración entre los Estados Partes
promueven el acceso de la población a medicamentos con mayor calidad y
seguridad.
Que el Acuerdo Nº 08/11 de la Reunión de Ministros de Salud del
MERCOSUR constituye un marco de referencia para la Farmacopea MERCOSUR.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar, en el marco de lo establecido en la Resolución GMC N°
22/14, el método general “Farmacopea MERCOSUR: Espectrofotometría
infrarrojo”, que consta como Anexo y forma parte de la presente
Resolución.
Art. 2 - Los Estados Partes indicarán en el ámbito del SGT N° 11 los
organismos nacionales competentes para la implementación de la presente
Resolución.
Art. 3 - Esta Resolución deberá ser incorporada al ordenamiento jurídico de los Estados Partes antes del 1/XII/2015.
XCVIII GMC - Brasilia, 29/V/15
ANEXO
ESPECTROFOTOMETRÍA INFRARROJO
Introducción
La radiación electromagnética es una forma de energía que se propaga
como ondas y puede ser subdividida en regiones de longitudes de onda
características. Asimismo, puede ser considerada también como un flujo
de partículas denominadas fotones (quanto). Cada fotón contiene
determinada energía cuya magnitud es proporcional a la frecuencia e
inversamente proporcional a la longitud de onda. La longitud de onda
(?) es generalmente especificada en nanómetros, nm (10-9 metros) y en
algunos casos en micrómetros, µm (10-6 metros). En caso del infrarrojo
la radiación electromagnética puede ser también descrita en términos de
número de onda y expresada en cm-1. Los rangos de longitud de onda de
energía electromagnética de interés para el infrarrojo se describen en
Tabla 1.
La espectrofotometría infrarroja es un método de medida de la absorción
de la radiación en un rango de longitudes de onda, cuando ésta pasa a
través de una capa delgada de sustancia.
La espectrofotometría de infrarrojo es un ensayo de identificación por
excelencia siendo capaz de distinguir sustancias con diferencias
estructurales. De las tres regiones de infrarrojo (cercano, medio y
lejano), la región comprendida entre 4000 a 400 cm-1 es la más empleada
para fines de identificación. Sin embargo, en algunos casos es
utilizado con fines cuantitativos. El espectro de infrarrojo (IR) es
único para cualquier compuesto químico con excepción de los isómeros
ópticos que tienen espectros idénticos. En algunas ocasiones, el
polimorfismo puede ser responsable de diferencias en el espectro IR de
un compuesto en estado sólido.
Debido al gran número de valores máximos en un espectro de absorción
IR, a veces es posible medir cuantitativamente los componentes
individuales de una mezcla con una composición cualitativa conocida sin
separación previa.
Cuando los ensayos por absorción infrarroja se aplican sobre una
muestra resultante de la extracción a partir de una formulación, puede
que no sea siempre posible una estricta concordancia con el espectro de
referencia. Sin embargo, el espectro del material extraído y el
espectro de referencia deberían alcanzar una similitud cercana. El
índice de concordancia deberá ser establecido en cada caso particular,
con su correspondiente verificación.
Equipo
Los espectrofotómetros utilizados para la obtención del infrarrojo
medio y cercano consisten de una fuente de luz, monocromador o
interferómetro y detector, los cuales permiten la obtención de
espectros en la región comprendida entre 780 nm a 25000 nm (12800 cm-1
a 400 cm-1). Actualmente, los espectrofotómetros de infrarrojo utilizan
un interferómetro en lugar de un monocromador en cuyo caso la radiación
policromática incide sobre la muestra y los espectros son obtenidos en
el dominio de la frecuencia con ayuda de la transformada de Fourier.
Verificación de desempeño del equipo
Escala de número de onda
La escala de número de onda puede ser verificada registrando el
espectro de un film de poliestireno que presente máximos de absorción a
los números de onda que se detallan a continuación:
Resolución
1- Para instrumentos que utilicen monocromador registrar el espectro de un film de poliestireno certificado de 35 µm de espesor.
2- Para instrumentos con Transformada de Fourier seleccionar una
resolución adecuada con una apodisación apropiada según indicación del
fabricante. Registrar el espectro de un film de poliestireno
certificado de 35 µm de espesor.
Especificaciones
Instrumentos con monocromador:
- La diferencia entre el porcentaje de transmitancia en el máximo de
transmisión a 2870 cm-1 (3,48 µm) y en el mínimo de transmisión a
2849,5 cm-1 (3,51 µm) debe ser mayor a 18.
- La diferencia entre el porcentaje de transmitancia en el máximo de
transmisión a 1589 cm-1 (6,29 µm) y en el mínimo de transmisión a 1583
cm-1 (6,32 µm) debe ser mayor a 10.
Instrumentos con transformada de Fourier:
- La diferencia entre las absorbancias en el mínimo de absorción a 2870
cm-1 y en el máximo de absorción a 2849,5 cm-1 debe ser mayor a 0,33.
- La diferencia entre las absorbancias en el mínimo de absorción a 1589
cm-1 y en el máximo de absorción a 1583 cm-1 debe ser mayor a 0,08.
Preparación de muestras y modos de medición
a) Método por medida de transmitancia o absorbancia
Se debe utilizar la muestra secada bajo las condiciones descritas en el
ensayo Perdida por secado, a no ser que se especifique de otra forma en
la monografía.
Se debe preparar la muestra de acuerdo a uno de los siguientes
procedimientos, según se indica en la monografía individual, de modo
que la transmitancia de la mayoría de las bandas se encuentre en el
rango de 5 a 80%.
- Método de disco de bromuro de potasio o cloruro de potasio (método en fase sólida)
Pulverizar 1 - 2 mg de la muestra sólida en un mortero de ágata,
triturar con 0,30 a 0,40 g de bromuro de potasio o cloruro de potasio
para espectrofotometría de infrarrojo, tomando precauciones respecto a
la absorción de humedad y comprimir la mezcla en un molde adecuado con
forma de disco. Si la sustancia a analizar es un clorhidrato, es
recomendable utilizar cloruro de potasio. Aplicar una presión de 50 a
100 kN por cm2 durante por lo menos 1 minuto, con ayuda de vacío en
caso de ser necesario, a fin de obtener un disco transparente de
aproximadamente 13 mm de diámetro.
El disco debe desecharse si no está uniformemente transparente cuando
se lo examina visualmente o si la transmitancia a 2.000 cm-1 (5 µm), en
ausencia de una banda de absorción específica, es menor de 75% sin
emplear compensación en el haz de referencia.
La humedad presente en la sustancia a analizar y/ o en la matriz
provocará apariencias irregulares en los discos de haluros de potasio,
tales como manchas y/ u opacidades.
Asimismo, y junto a la humedad aportada por el sistema analítico, ésta
contribuirá a la aparición de bandas en la región del IR cercano.
Por ello, es conveniente secar la sustancia a analizar bajo las
condiciones descriptas en el ensayo Pérdida por secado, a no ser que se
especifique de otra forma en la monografía.
Método para películas
Analizar una película delgada tal cual es o preparada como se describe en la monografía individual.
Método en solución para sólidos
Preparar la solución por el método indicado en la monografía
individual, en una celda para líquidos de material transparente a la
radiación infrarroja, y realizar la medida del espectro contra el
solvente de referencia utilizado para preparar la solución de la
sustancia a analizar. Normalmente, se obtienen resultados óptimos al
utilizar concentraciones en el rango de 10-100 g/L para caminos ópticos
de 0,1 mm a 0,5 mm. El solvente utilizado en este método no debería
exhibir interacciones o reacciones químicas con la sustancia a analizar
así como tampoco dañar la celda. Aquellas regiones del espectro en las
que el solvente presenta una fuerte absorción no deben tenerse en
cuenta.
Los solventes orgánicos a utilizar deben estar exentos de agua.
Método en suspensión para sólidos
Triturar 5 a 10 mg de la sustancia sólida a analizar con 2 gotas de
vaselina líquida u otro líquido apropiado hasta obtener una mezcla
cremosa homogénea. Colocar una porción de la mezcla así obtenida entre
dos placas de cloruro de sodio, bromuro de potasio u otro material
transparente a la radiación infrarroja y presionar suavemente las
placas para formar una película fina.
Método en película fina para líquidos
Colocar 1 o 2 gotas de la sustancia líquida a analizar entre dos placas
de cloruro de sodio, bromuro de potasio u otro material transparente a
la radiación infrarroja y presionar suavemente las placas para formar
una película fina.
Método en celda para líquidos
Puede emplearse una celda del mismo material descrito en Método en película fina para líquidos y de paso óptico apropiado.
Método para gases
Introducir la sustancia gaseosa a analizar en una celda para gases,
previamente evacuada y llenando a la presión que se especifica en la
monografía; analizar su espectro de absorción. La longitud del camino
óptico de la celda es usualmente 10 cm, pero si es necesario podría
exceder 1 m.
Para evitar interferencias de absorción debido al vapor de agua,
dióxido de carbono u otros gases atmosféricos, colocar en el haz de
referencia una celda idéntica evacuada o llenada con un gas
transparente a la radiación infrarroja, por ej. nitrógeno o argón. Si
es necesario, ajustar la presión en la celda a la presión atmosférica
usando un gas transparente a la radiación infrarroja (nitrógeno, o
argón).
b) Método de reflectancia difusa (DRIFT)
Método para sólidos
Preparar una mezcla compuesta por la sustancia a analizar y bromuro de
potasio o cloruro de potasio finamente pulverizados y secados, con una
concentración aproximada al 5%, a menos que se especifique de otro modo
en la monografía individual. Triturar en mortero de ágata con la
precaución de evitar la absorción de humedad, colocar la mezcla en el
accesorio correspondiente para sólidos, y registrar el espectro de
reflectancia.
Se recomienda que tanto la muestra como la matriz tengan un tamaño de partícula menor a 50 µm.
El espectro de reflectancia es tratado con el algoritmo de Kubelka -
Munk a fin de obtener el espectro en unidades de absorbancia.
c) Método reflectancia total atenuada (ATR)
Método para líquidos, geles, polvos, pastas, sólidos, películas y recubrimientos
Colocar la sustancia a ser analizada en estrecho contacto con un
elemento de reflectancia interna como diamante, germanio, seleniuro de
cinc, bromuro de talio- ioduro de talio (KRS-5) u otro material
apropiado de alto índice de refracción. Asegurar que el contacto entre
la sustancia a analizar y la totalidad de la superficie del elemento de
reflectancia interna sea uniforme, aplicando presión o disolviendo la
sustancia en un solvente apropiado mediante distribución sobre la
superficie del cristal y posterior evaporación hasta sequedad. Examinar
el espectro de ATR.
Criterios de identificación
Una sustancia puede ser identificada cuando presente únicamente máximos
de absorción en los mismos números de onda y con intensidades relativas
similares al espectro de una sustancia de referencia o al espectro de
referencia de la sustancia. Además cuando en la monografía son
especificadas absorciones a varios números de onda, la identificación
de la sustancia comparada con la sustancia esperada puede ser
confirmada por la presencia de las bandas de absorción a los números de
onda especificados.
A menos que se especifique de otra forma en la monografía individual,
se debe proceder mediante Identificación por uso de sustancia de
referencia.
1. Identificación por sustancia de referencia
Los máximos de absorción en el espectro obtenido con la muestra deben
corresponder en posición e intensidad relativa a los obtenidos
concomitantemente con la sustancia de referencia. En el caso de no
haber concordancia en los espectros de la muestra sólida y de la
sustancia de referencia, disolver porciones iguales de la muestra y de
la sustancia de referencia en volúmenes iguales de un solvente
apropiado, evaporar las soluciones hasta sequedad bajo condiciones
idénticas y repetir el ensayo con los residuos.
2. Identificación por espectro de referencia Farmacopea MERCOSUR
Preparar la muestra en condiciones similares a las indicadas para la
obtención del espectro de referencia y registrar el espectro de la
sustancia a analizar. Los máximos de absorción en el espectro obtenido
con la muestra deben corresponder en posición e intensidad relativa a
los exhibidos en el espectro de referencia.
En el caso de no haber concordancia entre el espectro obtenido con la
muestra sólida y el espectro de referencia, disolver la muestra en un
solvente apropiado, evaporar la solución hasta sequedad y repetir el
ensayo con el residuo.
Comparar los espectros y los máximos del poliestireno indicados en
Verificación de desempeño del equipo.
3. Identificación por máximos de absorción
Los máximos de absorción en el espectro obtenido con la muestra deben
corresponder en posición a todos los indicados en la monografía
individual.
MERCOSUL/GMC/RES. Nº 12/15
FARMACOPEA MERCOSUR: RANGO O TEMPERATURA DE FUSIÓN
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto y las Resoluciones N° 31/11 y 22/14 del Grupo Mercado Común.
CONSIDERANDO:
Que la Farmacopea MERCOSUR tiene como objetivo establecer los
requisitos mínimos de calidad y seguridad de los insumos para la salud,
especialmente de los medicamentos, apoyando las acciones de
reglamentación sanitaria y promoviendo el desarrollo técnico,
científico y tecnológico regional.
Que las especificaciones farmacopeicas establecen, por medio de
monografías, requisitos mínimos para el control de seguridad y calidad
de los insumos, especialidades farmacéuticas, plantas medicinales y
derivados producidos o utilizados en los Estados Partes.
Que las especificaciones farmacopeicas son utilizadas como parámetro
para las acciones de vigilancia sanitaria, incluyendo el registro de
medicamentos, inspecciones y análisis de laboratorio.
Que la Farmacopea MERCOSUR y la producción de patrones propios de
calidad favorecen al desarrollo científico y tecnológico de los Estados
Partes, contribuyendo a la disminución de la dependencia de proveedores
extranjeros y promoviendo a la industria regional.
Que la Farmacopea MERCOSUR debe ser primordialmente sanitaria, con
énfasis en la salud pública, y presentar una metodología analítica
accesible a los Estados Partes, buscando su reconocimiento y
respetabilidad internacional.
Que el diálogo regulatorio y la integración entre los Estados Partes
promueven el acceso de la población a medicamentos con mayor calidad y
seguridad.
Que el Acuerdo Nº 08/11 de la Reunión de Ministros de Salud del
MERCOSUR constituye un marco de referencia para la Farmacopea MERCOSUR.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar, en el marco de lo establecido en la Resolución GMC N°
22/14, el método general “Farmacopea MERCOSUR: rango o temperatura de
fusión”, que consta como Anexo y forma parte de la presente Resolución.
Art. 2 - Los Estados Partes indicarán en el ámbito del SGT N° 11 los
organismos nacionales competentes para la implementación de la presente
Resolución.
Art. 3 - Esta Resolución deberá ser incorporada al ordenamiento jurídico de los Estados Partes antes del 1/XII/2015.
XCVIII GMC - Brasilia, 29/V/15.
ANEXO
RANGO O TEMPERATURA DE FUSIÓN
La Temperatura o Punto de fusión de una sustancia se define como la
temperatura en la cual ésta se encuentra completamente fundida. Es una
propiedad intrínseca de las sustancias, la cual es utilizada, junto a
otros ensayos, para la confirmación de identidad de la misma; así como
indicador de pureza. En caso de sustancias que funden con
descomposición la temperatura o punto de fusión será la temperatura a
la cual comienza la fusión.
Rango de fusión de una sustancia se define como el rango comprendido
entre la temperatura en la cual la sustancia comienza a fluidificarse o
a formar gotas en las paredes del tubo capilar y la temperatura en la
cual la sustancia está completamente fundida.
Una transición de fusión puede ser instantánea para un material
altamente puro, pero por lo general se observa un intervalo desde el
comienzo hasta el final del proceso. Hay distintos factores que
influyen en esta transición y deben ser estandarizados cuando se
describe el procedimiento. Estos factores incluyen: cantidad de la
muestra, tamaño de partícula, eficiencia en la difusión del calor y la
velocidad del calentamiento entre otros.
Para fines farmacopeicos, el punto de fusión o rango de fusión se
informa como la temperatura a la cual se observa la primer fase líquida
y la temperatura a la cual no hay mas fase sólida aparente, excepto
aquellas sustancias que funden con descomposición o se especifique de
otra manera en la monografía individual.
Método 1
Para muestras que se reducen fácilmente a polvo.
Aparato I
Consta de un recipiente de vidrio (C) para un baño de líquido
transparente, un dispositivo mezclador (D), un termómetro (A), y una
fuente de calor adecuados (ver Figura). De acuerdo con la temperatura
requerida el líquido del baño puede ser uno de los siguientes u otro
que sea apropiado:
• Agua para temperaturas hasta 60 ºC
• Glicerina para temperaturas hasta 150 ºC
• Parafina líquida de alto rango de ebullición para temperaturas hasta 250 ºC
• Aceite de sésamo o un aceite siliconado de grado adecuado para temperaturas hasta 300 ºC
El líquido del baño debe tener la suficiente profundidad para permitir
la inmersión del termómetro a la profundidad especificada, de manera
que el bulbo quede aproximadamente a 2 cm del fondo del baño. El calor
puede ser suministrado por una llama o eléctricamente. El tubo capilar
tiene aproximadamente 10 cm de largo y entre 0,8 y 1,2 mm de diámetro
interno, con paredes de 0,1 a 0,3 mm de espesor y cerrado en uno de sus
extremos, a no ser que se especifique de otra forma en la monografía
individual. Se debe utilizar un dispositivo agitador que garantice la
homogeneidad de la temperatura en el baño.
Procedimiento
A menos que se especifique de otro modo en la monografía individual, proceder según se indica a continuación:
Reducir la muestra a polvo fino y secarla en un desecador al vacío sobre un agente desecante apropiado durante 24 horas.
Cargar el tubo capilar seco con suficiente cantidad de polvo hasta
formar una columna de 3 a 4 mm de altura, luego de haberlo comprimido
golpeándolo moderadamente sobre una superficie sólida. Unir el tubo
capilar al termómetro, ambos humedecidos con el líquido del baño.
Ajustar su altura, de modo que la muestra contenida en el capilar quede
junto al bulbo del termómetro (B).
Adaptar un termómetro auxiliar (E) de modo que el centro del bulbo
quede lo más cercano posible al vástago del termómetro principal (A) en
un punto equidistante de la superficie del baño y de la división
correspondiente al punto de fusión esperado.
Calentar el baño hasta alcanzar una temperatura de 10 °C por debajo del
punto de fusión esperado. Introducir el termómetro con el capilar
adherido y continuar el calentamiento de manera tal que la temperatura
se eleve a una velocidad de 1 a 2 ºC por minuto, dependiendo de la
estabilidad de la sustancia.
Registrar la lectura del termómetro auxiliar al terminar la fusión de
la muestra, si fuera necesario, aplicar la corrección por columna
emergente empleando la fórmula siguiente:
Realizar la determinación al menos por triplicado. Para ello, dejar
enfriar el baño hasta 10 ºC por debajo del punto de fusión o hasta una
temperatura más baja y repetir el procedimiento empleando nuevas
porciones de muestra.
Aparato II
Consta de un bloque metálico que puede ser calentado a velocidad
controlada, cuya temperatura puede ser monitoreada por un sensor o
termómetro. El bloque permite colocar el tubo capilar que contiene la
sustancia en ensayo y monitorear el proceso de fusión mediante control
visual o automáticamente.
Procedimiento
A menos que se especifique de otro modo en la monografía individual, proceder según se indica a continuación:
Reducir la muestra a polvo fino y secarla en un desecador al vacío sobre un agente desecante apropiado durante 24 horas.
Cargar el tubo capilar seco con suficiente cantidad de polvo hasta
formar una columna de 3 a 4 mm de altura, luego de haberlo comprimido
golpeándolo moderadamente sobre una superficie sólida.
Calentar el bloque rápidamente hasta una temperatura de 10 ºC por
debajo del punto de fusión esperado y continuar el calentamiento de
manera tal que la temperatura se eleve a una velocidad de 1 a 2 °C por
minuto. Introducir el capilar en el bloque y registrar la temperatura
al inicio y fin de la fusión.
Realizar la determinación al menos por triplicado. Para ello, dejar
enfriar el bloque hasta 10 °C por debajo del punto de fusión o hasta
una temperatura más baja y repetir el procedimiento empleando nuevas
porciones de muestra.
Método 2
Para muestras que no se reducen fácilmente a polvo.
Procedimiento
Fundir cuidadosamente la muestra a la temperatura más baja posible e
introducir el material fundido en un capilar abierto en ambos extremos,
hasta formar una columna de unos 10 mm de altura. Enfriar el capilar
cargado a una temperatura menor o igual a 10 ºC durante aproximadamente
24 horas. Unir el capilar al termómetro y ajustar su altura, de modo
que la muestra contenida en el capilar quede junto al bulbo del
termómetro. Introducir en un baño de agua y calentar según se indica en
Método 1 Aparato I, excepto que al llegar a una temperatura
aproximadamente de 5 ºC por debajo del punto de fusión esperado, se
aumenta la temperatura a una velocidad de 0,5 °C por minuto. Se toma
como punto de fusión la temperatura a la cual la muestra comienza a
ascender dentro del tubo capilar. Realizar la determinación al menos
por triplicado utilizando porciones diferentes de muestra.
Método 3
Para vaselina, sustancias grasas u otras de consistencia pastosa.
Procedimiento
Fundir la muestra, agitando hasta alcanzar una temperatura de 90 a 92
ºC y luego dejar enfriar la sustancia fundida hasta una temperatura de
8 a 10 ºC sobre el punto de fusión esperado. Enfriar hasta 5 ºC el
bulbo del termómetro, secar y, mientras esté aún frío, sumergirlo en la
muestra fundida hasta la mitad del bulbo aproximadamente. Retirar
inmediatamente y sostener en posición vertical, hasta que la superficie
de la muestra depositada sobre el bulbo solidifique. Introducir en un
baño de agua a una temperatura que no exceda los 16 ºC durante 5
minutos aproximadamente.
Adaptar el termómetro dentro de un tubo de ensayo, por medio de un
tapón perforado, de modo que su extremo inferior quede 15 mm por encima
del fondo del tubo. Suspender el tubo de ensayo en un baño de agua a
una temperatura de 16 ºC y elevar la temperatura del baño hasta 30 ºC,
a una velocidad de 2 ºC por minuto, y luego a una velocidad de 1 ºC por
minuto hasta que la primera gota se desprenda del termómetro. La
temperatura a la cual esto sucede es el punto de fusión. Para cada
determinación emplear una porción recién fundida de la muestra.
Realizar la determinación por triplicado. Si la máxima diferencia entre
las determinaciones es menor de 1 ºC, promediar los valores obtenidos.
De lo contrario, realizar dos determinaciones adicionales y promediar
los cinco.
MERCOSUR/GMC/RES. Nº 13/15
FARMACOPEA MERCOSUR: PÉRDIDA POR SECADO
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto y las Resoluciones N° 31/11 y 22/14 del Grupo Mercado Común.
CONSIDERANDO:
Que la Farmacopea MERCOSUR tiene como objetivo establecer los
requisitos mínimos de calidad y seguridad de los insumos para la salud,
especialmente de los medicamentos, apoyando las acciones de
reglamentación sanitaria y promoviendo el desarrollo técnico,
científico y tecnológico regional.
Que las especificaciones farmacopeicas establecen, por medio de
monografías, requisitos mínimos para el control de seguridad y calidad
de los insumos, especialidades farmacéuticas, plantas medicinales y
derivados producidos o utilizados en los Estados Partes.
Que las especificaciones farmacopeicas son utilizadas como parámetro
para las acciones de vigilancia sanitaria, incluyendo el registro de
medicamentos, inspecciones y análisis de laboratorio.
Que la Farmacopea MERCOSUR y la producción de patrones propios de
calidad favorecen al desarrollo científico y tecnológico de los Estados
Partes, contribuyendo a la disminución de la dependencia de proveedores
extranjeros y promoviendo a la industria regional.
Que la Farmacopea MERCOSUR debe ser primordialmente sanitaria, con
énfasis en la salud pública, y presentar una metodología analítica
accesible a los Estados Partes, buscando su reconocimiento y
respetabilidad internacional.
Que el diálogo regulatorio y la integración entre los Estados Partes
promueven el acceso de la población a medicamentos con mayor calidad y
seguridad.
Que el Acuerdo Nº 08/11 de la Reunión de Ministros de Salud del
MERCOSUR constituye un marco de referencia para la Farmacopea MERCOSUR.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar, en el marco de lo establecido en la Resolución GMC N°
22/14, el método general “Farmacopea MERCOSUR: Pérdida por secado”, que
consta como Anexo y forma parte de la presente Resolución.
Art. 2 - Los Estados Partes indicarán en el ámbito del SGT N° 11 los
organismos nacionales competentes para la implementación de la presente
Resolución.
Art. 3 - Esta Resolución deberá ser incorporada al ordenamiento jurídico de los Estados Partes antes del 1/XII/2015.
XCVIII GMC - Brasilia, 29/V/15.
ANEXO
PÉRDIDA POR SECADO
Este ensayo se destina a determinar la cantidad de sustancia volátil de
cualquier naturaleza eliminada en las condiciones especificadas en la
monografía individual. Para sustancias que tienen agua como único
constituyente volátil es apropiado aplicar el procedimiento que se
indica en el capítulo Determinación de Agua. El resultado se expresa en
porcentaje p/p, calculado de la siguiente manera:
Procedimiento
1. Gravimetría
A menos que se especifique de otra manera en la monografía individual proceder según se indica a continuación:
En caso de ser necesario, reducir la sustancia a polvo fino triturando rápidamente.
Pesar una cantidad aproximada entre 1 - 2 g de sustancia, de forma
exacta, en un pesa-filtro previamente secado durante 30 minutos, en las
mismas condiciones que son empleadas en el ensayo de la muestra, y
enfriado a temperatura ambiente en un desecador.
Distribuir la muestra lo más uniformemente posible, agitando suavemente
el pesa-filtro de modo que se forme una capa de 5 mm de espesor
aproximadamente y no más de 10 mm en el caso de materiales voluminosos.
Colocar el pesa-filtro conteniendo la muestra, destapado, junto con la
tapa en la cámara de secado. Secar la muestra en las condiciones
especificadas en la monografía. (Nota: la temperatura especificada en
la monografía debe considerarse comprendida en el intervalo de ± 2 ºC).
Abrir la cámara de secado, tapar el pesa-filtro rápidamente, retirarlo
y permitir que llegue a temperatura ambiente en un desecador antes de
pesarlo.
Cuando en la monografía individual se especifica secar hasta peso
constante el secado deberá continuarse hasta que dos pesadas
consecutivas no difieran en más de 0,50 mg por gramo de sustancia
tomada, realizando la segunda pesada después de una hora adicional de
secado.
Si la sustancia funde a una temperatura inferior a la que se especifica
para la determinación de la pérdida por secado mantener el pesa-filtro
con su contenido durante 1 a 2 horas a una temperatura de 5 a 10 ºC
inferior a la temperatura de fusión y luego secar a la temperatura
especificada.
Para el análisis de cápsulas utilizar una porción de contenido
homogeneizado de no menos de 4 unidades. En el caso de comprimidos
utilizar el polvo de no menos de 4 unidades.
Cuando en la monografía individual indique:
• secado al vacío se deberá utilizar un desecador al vacío, una estufa de secado al vacío u otro aparato adecuado;
• secar al vacío en un frasco con tapón provisto de perforación
capilar, se deberá utilizar un frasco o tubo con un tapón capilar de
225 ± 25 µm de diámetro y mantener la cámara de calentamiento a una
presión de 5 mm de mercurio o menor. Al final del período de
calentamiento, dejar entrar aire seco a la cámara, retirar el frasco y
con el tapón con perforación capilar todavía en su sitio, permitir que
se enfríe a temperatura ambiente en un desecador antes de pesar;
• secado en un desecador, se deberán tener las precauciones necesarias
para asegurar que el agente desecante se mantenga activo. Dentro de los
agentes desecantes más frecuentes se encuentran cloruro de calcio,
sílica gel y pentóxido de fósforo.
2. Termogravimetría
En caso que la monografía individual especifique que la pérdida por
secado debe realizarse por análisis termogravimétrico, proceder como
especifica el capítulo Análisis Térmico.
3. Balanza infrarrojo o con lámpara halógena
En caso que la monografía individual especifique que la pérdida por
secado debe realizarse con balanza infrarrojo o con lámpara halógena
proceder según se indica a continuación:
• Retirar la humedad del equipo.
• Pesar la cantidad de sustancia a ser analizada y distribuir
uniformemente el material en el colector de muestra y colocarlo dentro
del aparato.
• Definir el tiempo y la temperatura de secado según lo establezca la
monografía individual. Registrar el valor de humedad obtenido.
MERCOSUR/GMC/RES. Nº 14/15
FARMACOPEA MERCOSUR: VACUNAS DE USO HUMANO
VISTO: El Tratado de Asunción, el Protocolo de Ouro Preto y las Resoluciones N° 31/11 y 22/14 del Grupo Mercado Común.
CONSIDERANDO:
Que la Farmacopea MERCOSUR tiene como objetivo establecer los
requisitos mínimos de calidad y seguridad de los insumos para la salud,
especialmente de los medicamentos, apoyando las acciones de
reglamentación sanitaria y promoviendo el desarrollo técnico,
científico y tecnológico regional.
Que las especificaciones farmacopeicas establecen, por medio de
monografías, requisitos mínimos para el control de seguridad y calidad
de los insumos, especialidades farmacéuticas, plantas medicinales y
derivados producidos o utilizados en los Estados Partes.
Que las especificaciones farmacopeicas son utilizadas como parámetro
para las acciones de vigilancia sanitaria, incluyendo el registro de
medicamentos, inspecciones y análisis de laboratorio.
Que la Farmacopea MERCOSUR y la producción de patrones propios de
calidad favorecen al desarrollo científico y tecnológico de los Estados
Partes, contribuyendo a la disminución de la dependencia de proveedores
extranjeros y promoviendo a la industria regional.
Que la Farmacopea MERCOSUR debe ser primordialmente sanitaria, con
énfasis en la salud pública, y presentar una metodología analítica
accesible a los Estados Partes, buscando su reconocimiento y
respetabilidad internacional.
Que el diálogo regulatorio y la integración entre los Estados Partes
promueven el acceso de la población a medicamentos con mayor calidad y
seguridad.
Que el Acuerdo Nº 08/11 de la Reunión de Ministros de Salud del
MERCOSUR constituye un marco de referencia para la Farmacopea MERCOSUR.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar, en el marco de lo establecido en la Resolución GMC N°
22/14, la monografía “Farmacopea MERCOSUR: Vacunas de uso humano”, que
consta como Anexo y forma parte de la presente Resolución.
Art. 2 - Los Estados Partes indicarán en el ámbito del SGT N° 11 los
organismos nacionales competentes para la implementación de la presente
Resolución.
Art. 3 - Esta Resolución deberá ser incorporada al ordenamiento jurídico de los Estados Partes antes del 1/XII/2015.
XCVIII GMC - Brasilia, 29/V/15.
ANEXO
VACUNAS DE USO HUMANO
Vaccina ad usum humanuno
DEFINICIÓN
Las vacunas para uso humano son preparaciones que contienen sustancias
antigénicas capaces de inducir en el hombre una inmunidad activa
específica contra un agente infeccioso. Su eficacia y seguridad deben
ser demostradas mediante estudios aprobados por una Autoridad
Regulatoria Nacional competente.
Las vacunas para uso humano pueden estar compuestas por microorganismos
inactivados, microorganismos vivos atenuados, antígenos extraídos o
secretados de microorganismos o producidos por ingeniería genética.
PRODUCCIÓN
Consideraciones Generales
Los requisitos para la producción así como los controles en proceso
están incluidos en las monografías individuales. Los procedimientos de
preparación deben obedecer a normas de Buenas Prácticas de Fabricación
de productos farmacéuticos.
Durante el proceso de producción de vacunas pueden ser incorporados
sustancias como estabilizantes, adyuvantes y conservantes. Los
componentes que se conozcan causen reacciones tóxicas, alérgicas y
otros efectos indeseables en el hombre no pueden usarse.
No pueden utilizarse penicilina ni estreptomicina ni sus derivados en
ninguna etapa de la producción ni pueden ser agregadas al producto
final. Siempre que se justifique y autorice su uso se pueden utilizar
en el lote semilla primario.
Para la utilización de albúmina en el proceso de producción, la misma
debe cumplir con los requisitos descriptos en la monografía de Solución
de Albúmina Humana descritas en las normativas nacionales.
En la producción de vacunas, para el número de pasajes o subcultivos,
se debe considerar los ensayos clínicos utilizados para demostrar la
seguridad y eficacia de la vacuna.
PRODUCTO FINAL
En los productos liofilizados como en las preparaciones líquidas
monodosis no se acepta la utilización de conservantes antimicrobianos.
En el caso de las preparaciones líquidas multidosis se evaluará en cada
caso la necesidad de la utilización de conservantes antimicrobianos.
Cuando se utiliza suero de origen animal en el proceso de producción, el producto final no puede contener más de 50 ng/dosis.
IDENTIFICACIÓN
Debe cumplir con el ensayo descrito en la monografía individual.
CARACTERIZACIÓN
Debe cumplir con el(los)ensayo(s) descrito(s) en la monografía individual, cuando sea aplicable.
ENSAYOS FISICO-QUIMICOS
Debe cumplir con el(los) ensayo(s) descrito(s) en monografía individual.
ENSAYOS DE PUREZA
Debe cumplir con el(los) ensayo(s) descrito(s) en monografía individual.
ENSAYOS BIOLÓGICOS Y MICROBIOLÓGICOS
Debe cumplir con el(los) ensayo(s) descrito(s) en monografía individual.
POTENCIA
Debe cumplir con el ensayo descrito en monografía individual.
TERMOESTABILIDAD
Debe cumplir con el ensayo descrito en la monografía individual, cuando sea aplicable.
ACONDICIONAMIENTO Y ALMACENAMIENTO
De acuerdo con la normativa vigente aprobada por la Autoridad Regulatoria Nacional.
ROTULADO
De acuerdo con la normativa vigente aprobada por la Autoridad Regulatoria Nacional.
GLOSARIO
Adyuvante de inmunización
Los adyuvantes son sustancias o combinación de ellas que incorporados
al antígeno o inyectados simultáneamente con él, aumentan la respuesta
inmune específica y eficacia clínica de la vacuna.
Agentes adventicios
Microorganismos contaminantes de cultivos celulares o materias primas,
incluyendo bacterias, hongos, micoplasmas, micobacterias y virus
endógenos o exógenos que puedan ser introducidos durante el proceso de
fabricación de un producto biológico.
Banco de células
Cultivo de células distribuidas en envases de composición uniforme y
derivados de un único conjunto de células. Se almacena criogénicamente
bajo condiciones definidas que garantizan su estabilidad.
Banco de células maestro
Cantidad de células bien caracterizadas, obtenidas de una única célula
totalmente definida, de origen animal u otro, distribuida en
recipientes en una única operación. Posee composición uniforme,
criopreservado y almacenados bajo condiciones definidas.
Banco de células de trabajo
Cultivo de células preparado a partir del banco maestro de células bajo
condiciones de cultivo definidas, almacenado criogénicamente y usado
para iniciar el cultivo de células en la producción.
Células de crecimiento continuo
Una línea celular que aparentemente tenga capacidad ilimitada para duplicarse.
Cosecha única
Suspensión de microrganismos obtenida a partir de un cultivo inoculado
con el lote de trabajo o con el inóculo de producción, recogida y
procesada en un ciclo único de producción.
Cultivo celular de producción
Usado en la producción de productos biológicos y derivado de uno o más
envases del banco de células de trabajo, o en el caso de cultivos
celulares primarios, de los tejidos de animales.
Cultivo primario
Cultivo iniciado de células, tejidos u órganos provenientes
directamente de uno o más organismos. Un cultivo puede ser considerado
primario hasta que sea subcultivado satisfactoriamente la primera vez.
Luego se transforma en una “línea celular” si se puede continuar
subcultivando varias veces.
Estándar primario
Son muestras de fármacos, impurezas, productos de degradación,
reactivos, entre otros, altamente caracterizados y de la más elevada
pureza, cuyo valor se acepta sin referencia a otros estándares.
Estándar secundario (estándar de trabajo)
Estándar utilizado en la rutina del laboratorio, cuyo valor es establecido por comparación con un estándar de referencia.
Inóculo de producción
Suspensión de microorganismos, de composición uniforme, obtenida a partir de una o más ampollas del lote de trabajo.
Línea celular
Tipo de población celular con características definidas originaria de
subcultivos de una población de célula primaria. Puede generarse
mediante la utilización de etapas de clonado y subclonado.
Línea de célula diploide
Línea celular con vida útil limitada in vitro, con los cromosomas
apareados (euploide) y estructuralmente idénticos a aquellos de las
especies de las cuales derivan.
Lote semilla primario
Ampollas conteniendo microorganismos conservados de composición
uniforme, obtenida a partir de una cepa conservada de procedencia
conocida.
Lote semilla de trabajo
Ampollas conteniendo microorganismos conservados de composición
uniforme obtenida a partir de un lote de semilla primario y destinado a
ser utilizado en la producción.
Polisacárido purificado
Material polisacárido obtenido después de la purificación final. El
lote de polisacárido purificado puede provenir de una cosecha
individual o de una mezcla de cosechas individuales procesadas en un
mismo ciclo.
Polisacárido modificado
Polisacárido purificado que se ha modificado por reacción química o
proceso físico en la preparación para conjugación a una proteína
transportadora.
Producto biológico
Medicamento de origen biológico, que contenga molécula con actividad
biológica demostrada, empleado para la prevención, diagnóstico y/o
tratamiento de una enfermedad o estado patológico y cuya calidad,
seguridad y eficacia han sido demostradas.
Producto biológico a granel
Producto biológico que ha completado todas las etapas de producción,
formulado en su forma farmacéutica final a granel contenido en un
recipiente único, estéril si es aplicable, y liberado por el control de
calidad del fabricante.
Producto biológico en su envase primario
Producto biológico que haya completado todas las etapas de producción,
formulado en su forma farmacéutica final, contenido en su envase final,
estéril, si fuera necesario, liberado por el control de calidad del
fabricante, sin incluir el proceso de rotulado y empaque.
Producto biológico intermedio
Producto farmacéutico, de origen biológico, parcialmente procesado,
susceptible de especificación y cuantificación, que será sometido a las
etapas subsiguientes de formulación / fabricación, antes de convertirse
en un producto a granel.
Producto biológico terminado
Producto farmacéutico, de origen biológico, que haya completado todas
las fases de producción, incluyendo el rotulado y empaque final.
Producto biotecnológico
Producto farmacéutico, de origen biológico, cuyo principio activo se
obtiene por un proceso del ADN recombinante, hibridomas, líneas
celulares transformadas u otras técnicas de biotecnológicas, con
finalidades profilácticas, curativas, paliativas o con fines de
diagnóstico in vivo.
Proteína transportadora
Proteína en la que el polisacárido está unido covalentemente con el
objetivo de inducir respuesta inmune dependiente de células T contra el
polisacárido.
Sustrato celular
Células utilizadas para fabricar un producto biológico. Pueden ser
células primarias o de línea celular, cultivadas en condiciones de
monocapas o suspensiones.
Toxoides bacterianos
Los toxoides bacterianos son toxinas detoxificadas por tratamientos
fisicoquímicos, que a pesar de perder su capacidad tóxica, mantienen la
actividad inmunogénica.
Vacunas bacterianas
Las vacunas bacterianas son producidas en medios líquidos o sólidos,
utilizando cepas adecuadas y contienen bacterias inactivadas, bacterias
atenuadas o sus componentes antigénicos.
Vacunas combinadas
Las vacunas combinadas contienen una mezcla de dos o más antígenos
diferentes. Pueden contener en su formulación, microorganismos
atenuados o inactivados, sustancias producidas por ellos y fracciones
antigénicas.
Vacunas conjugadas
Las vacunas conjugadas son aquellas en las cuales los antígenos
bacterianos están unidos a proteínas transportadoras, facilitando el
reconocimiento por los linfocitos T, generando una respuesta de
anticuerpos de larga duración.
Vacunas virales
Las vacunas virales consisten en preparaciones de virus enteros
atenuados, inactivados o por sus fracciones. Los virus pueden ser
replicados en animales, huevos embrionados, líneas celulares
susceptibles, o en un vector de expresión específico.
“El Acta de la XCVIII Reunión Ordinaria del Grupo Mercado Común (GMC),
realizada entre los días 28 y 29 de mayo de 2015 en la ciudad de
Brasilia y los anexos mencionados en la misma se encuentran a
disposición del público en el sitio web Oficial del MERCOSUR
(www.mercosur.int.) de conformidad con lo que establece el Protocolo de
Ouro Preto aprobado por Ley 24.560.”
Ing. MARCELO O. MARZOCCHINI, Director de Asuntos Económicos del MERCOSUR.
NOTA: El/los Anexo/s que integra/n este(a) Aviso Oficial se publican en
la edición web del BORA —www.boletinoficial.gov.ar— y también podrán
ser consultados en la Sede Central de esta Dirección Nacional (Suipacha
767 - Ciudad Autónoma de Buenos Aires).
e. e. 07/08/2015 N° 132130/15 v. 07/08/2015
(Nota Infoleg:
Los anexos referenciados en la presente norma han sido extraídos de la
edición web de Boletín Oficial. Los mismos pueden consultarse en el
siguiente link: Anexos)