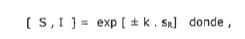MINISTERIO DE SALUD
SECRETARÍA DE POLÍTICAS, REGULACIÓN E INSTITUTOS
ADMINISTRACIÓN NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGÍA MÉDICA
Disposición 12704/2016
Buenos Aires, 15/11/2016
VISTO la Ley N° 16.463 y los Decretos Reglamentarios Nros. 9763/64 y
150/92 (T.O. 1993), 1490/92 y sus modificatorios, las Disposiciones
ANMAT N° 3185/99 y complementarias, 5040/06, 1746/07, 556/09, 758/09,
4132/12, 4133/12, 4326/12, 4788/12, 1918/13 y 6677/10 y el Expediente
N° 1-47-0000-013615-16-1 del Registro de esta Administración Nacional
de Medicamentos, Alimentos y Tecnología Médica; y
CONSIDERANDO:
Que el artículo 1° de la Ley 16.463 establece que “quedan sometidos a
la presente ley y a los reglamentos que en su consecuencia se dicten,
la importación, exportación, producción, elaboración, fraccionamiento,
comercialización o depósito en jurisdicción nacional o con destino al
comercio interprovincial de las drogas, productos químicos, reactivos,
formas farmacéuticas, medicamentos, elementos de diagnóstico, y todo
otro producto de uso y aplicación en medicina humana y las personas de
existencia visible o ideal que intervengan en dichas actividades”.
Que el artículo 2° de la citada ley establece que las actividades
mencionadas sólo podrán realizarse previa autorización y bajo el
contralor de la autoridad sanitaria, en establecimientos por ella
habilitados y bajo la dirección técnica del profesional universitario
correspondiente; todo ello en las condiciones y dentro de las normas
que establezca la reglamentación, atendiendo a las características
particulares de cada actividad y a razonables garantías técnicas en
salvaguarda de la salud pública y de la economía del consumidor.
Que asimismo el artículo 3° del mencionado cuerpo legal prescribe que
los productos comprendidos en la citada ley deberán reunir las
condiciones establecidas en la Farmacopea Argentina, y en caso de no
figurar en ella, las que surgen de los patrones internacionales y de
los textos de reconocido valor científico, debiendo a la vez ser
inscriptos por ante esta Administración Nacional de conformidad a lo
establecido en el Decreto N° 150/92 (T.O. 1993).
Que el artículo 1° del Decreto N° 9763/64, reglamentario de la Ley
16463, establece que el ejercicio del poder de policía sanitaria
referido a las actividades indicadas en el artículo 1° de dicha ley y a
las personas de existencia visible o ideal que intervengan en las
mismas, se hará efectivo por el entonces Ministerio de Asistencia
Social y Salud Pública de la Nación, hoy Ministerio de Salud, en las
jurisdicciones que allí se indican.
Que por su parte el Decreto N° 1490/92, crea esta Administración
Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT), en el
ámbito del Ministerio de Salud como organismo descentralizado de la
Administración Pública Nacional, con un régimen de autarquía financiera
y económica, con jurisdicción en todo el territorio nacional, asumiendo
dichas funciones.
Que por Disposición ANMAT N° 3185/99 se establecieron las exigencias de
estudios de biodisponibilidad/bioequivalencia entre productos y se
adoptó el criterio para su implementación gradual de acuerdo al riesgo
sanitario de su Ingrediente Farmacéutico Activo (IFA).
Que las Disposiciones ANMAT N° 3185/99 y 5040/06 establecen los límites
80% - 125% del intervalo de confianza 90% para la razón de las medias
geométricas del área bajo la curva (AUC) y la concentración máxima
(Cmax) para aceptar la bioequivalencia entre un producto multifuente y
el producto comparador de referencia.
Que la Disposición ANMAT N° 1746/07 establece como criterios de
aceptación de bioequivalencia, además de los descriptos, límites de
bioequivalencia ampliados para la Cmax siempre que no produzcan
modificaciones en la eficacia y seguridad del medicamento.
Que se han identificado IFAs y formulaciones cuya variabilidad
intrasujeto presenta un coeficiente de variación (CV) igual o mayor al
30% en los parámetros Cmax y AUC, y han sido categorizados como IFAs de
alta variabilidad.
Que la variabilidad intrasujeto puede definirse como una medida de la
variabilidad en la respuesta en el mismo sujeto cuando se le administra
la misma dosis de la especialidad medicinal en dos períodos diferentes.
Que los IFAS de alta variabilidad intrasujeto poseen en general alguna
de las siguientes características farmacocinéticas y farmacodinámicas:
amplio margen terapéutico, baja concentración en plasma debido a
extenso metabolismo pre-sistémico, alta labilidad en medio ácido, alta
lipofilicidad.
Que de acuerdo a la Clasificación Biofarmacéutica, los IFAs de alta
variabilidad se categorizan en general como IFAs de Clase II (alta
permeabilidad, baja solubilidad) y de Clase IV (baja permeabilidad,
baja solubilidad).
Que han sido identificados IFAs de distinta categoría terapéutica que
poseen alta variabilidad intrasujeto como los inhibidores de la enzima
convertidora de la angiotensina, los inhibidores de la reductasa de 3
hidroxi-3-metilglutaril-coenzima A o los antagonistas del receptor
angiotensina II.
Que determinar la bioequivalencia de IFAs o formulaciones de alta
variabilidad resulta un reto dado que la alta variabilidad intrasujeto
significa que puede ser necesario incorporar al estudio un importante
número de sujetos para alcanzar un adecuado poder estadístico y
ajustarse al límite de bioequivalencia pre-determinado 80% - 125%.
Que con la finalidad de limitar el tamaño muestral necesario para la
demostración de bioequivalencia de IFAs o formulaciones de alta
variabilidad intrasujeto, otras agencias regulatorias como la EMA y la
FDA y como así también organizaciones no gubernamentales como la OMS
han adoptado los criterios de bioequivalencia promedio con escalamiento
al producto de referencia.
Que en consonancia con pautas internacionales la ANMAT desaconseja la
realización innecesaria de ensayos en humanos por lo que surge la
controversia si resulta necesario incluir un elevado número de sujetos
en los estudios de bioequivalencia para los cuales la alta variabilidad
no parece impactar en la seguridad y eficacia.
Que el elevado número muestral de voluntarios sanos necesario para
estudios de bioequivalencia con IFAs o formulaciones de alta
variabilidad, ha llevado a examinar distintas alternativas al uso de
los límites 80% - 125% del intervalo de confianza 90% para la razón de
las medias geométricas del AUC y Cmax, definido como bioequivalencia
promedio.
Que la bioequivalencia promedio declara bioequivalentes a aquellas
formulaciones cuya diferencia de medias µM - µR pertenece a un
intervalo prefijado (?1, ?2) llamado intervalo de bioequivalencia, lo
que implica verificar la condición: ?1 < µM - µR < ?2 donde µM es
el valor esperado para la muestra que recibió la formulación
multifuente y µR es el valor esperado para la muestra que recibió la
formulación de referencia.
Que la bioequivalencia promedio con escalamiento al producto de
referencia significa ajustar el intervalo de bioequivalencia a la
variabilidad intrasujeto del producto de referencia determinado a
través de un diseño replicativo.
Que por todo lo mencionado corresponde establecer las recomendaciones
para la aplicación de la ampliación de los límites de aceptación para
los IFAs o formulaciones de alta variabilidad intrasujeto.
Que el Instituto Nacional de Medicamentos, la Dirección de
Fiscalización y Gestión de Riesgo, la Dirección de Registro y
Evaluación de Medicamentos y la Dirección General de Asuntos Jurídicos,
han tomado la intervención de su competencia.
Que se actúa en virtud de las facultades conferidas por el Decreto N°
1490 de fecha 20 de agosto de 1992 y el Decreto Nº 101 del 16 de
diciembre de 2015.
Por ello;
EL ADMINISTRADOR NACIONAL DE LA ADMINISTRACIÓN NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGÍA MÉDICA
DISPONE:
ARTÍCULO 1° — Adóptense los criterios de la bioequivalencia promedio
con escalamiento al producto de referencia para los IFAs o
formulaciones de alta variabilidad intrasujeto para el parámetro
farmacocinético concentración máxima (Cmax) siempre que dicho
escalamiento no tenga ningún impacto en la seguridad y eficacia del
producto. Para la aceptación de los mencionados criterios el estudio de
bioequivalencia deberá seguir un diseño replicado y la variabilidad
intrasujeto para el parámetro Cmax del producto de referencia deberá
ser mayor al 30%.
ARTÍCULO 2° — La determinación de la amplitud del intervalo de
aceptación de bioequivalencia del parámetro Cmax es definida de acuerdo
a la ecuación descripta en al Anexo I de la presente disposición que
forma parte integrante de la misma.
ARTÍCULO 3° — Establécese que la razón de la media geométrica del
parámetro Cmax deberá situarse dentro del intervalo 80% - 125%.
ARTÍCULO 4° — La ampliación de los criterios de aceptación basados en
la variabilidad intrasujeto no será aplicable al parámetro AUC para el
que se mantendrá el intervalo 80% - 125% sin considerar la variabilidad.
ARTÍCULO 5° — Adóptase la tabla de límites de aceptación para los
diferentes niveles de variabilidad que figura en el Anexo II de la
presente disposición y forma parte integrante de la misma.
ARTÍCULO 6° — La presente disposición entrará en vigencia el día siguiente al de su publicación en el Boletín Oficial.
ARTÍCULO 7° — Regístrese; Dése a la Dirección Nacional del Registro
Oficial para su publicación. Comuníquese a la Confederación
Farmacéutica de la República Argentina (COFA), a la Federación
Argentina de Cámaras de Farmacia (FACAF), a la Cámara de Farmacias, a
CILFA, CAEME, COOPERALA y CAPGEN. Cumplido, archívese. — Dr. CARLOS
CHIALE, Administrador Nacional, A.N.M.A.T.
ANEXO I
La amplitud del intervalo de bioequivalencia estará sujeta a la siguiente ecuación:
S: Límite superior del intervalo de aceptación
I: Límite inferior del intervalo de aceptación.
Constante cuyo valor es 0.760
SR: Desviación estándar intrasujeto del parámetro log-transformado concentración máxima (Cmax) del producto de referencia.
ANEXO II
Tabla 1. Límites de aceptación para los diferentes niveles de variabilidad
| Coeficiente de Variación |
LÍMITE |
| Intrasujeto (%) |
Inferior |
Superior |
| 30 |
80.00 |
125.00 |
| 35 |
77.23 |
129.48 |
| 40 |
74.62 |
134.02 |
| 45 |
72.15 |
138.59 |
| = 50 |
69.84 |
143.19 |
e. 16/11/2016 N° 87208/16 v. 16/11/2016