ENFERMEDADES
INCLUIDAS EN EL SISTEMA ÚNICO DE REINTEGRO POR GESTIÓN DE ENFERMEDADES
(SURGE)
Contenido

Los requisitos de empadronamiento y seguimiento de los pacientes con
patología oncológica incluida para apoyo financiero resultan comunes
para todos los subtipos detallados en el presente anexo. Los
fundamentos terapéuticos específicos son listados según el tipo de
Cáncer.
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Estadio Inicial (I, II, III, IV)
3. Matrícula del Profesional Tratante
4. ECOG (Eastern Cooperative Oncology
Group)
5. Fecha de última recaída o progresión
6. Sitio de Metástasis (Hígado,
Pulmón, SNC, Ganglionar, Óseo, Serosas, otros)
7. Tratamientos Previos (según tipo de
Cáncer)
8. Fecha de inicio de tratamiento
sujeto a recupero
9. Esquema de quimioterapia
concomitante
10. Estrategia - Línea terapéutica
o Adyuvancia-Neoadyuvancia
o Primera línea estadio avanzado
o Segunda línea estadio avanzado
o Tercera línea estadio avanzado
11. Documentación Respaldatoria
o Resumen de Historia Clínica (debe
detallar antecedentes, estadificación, terapias previas, estado actual
y estatus funcional)
o Estadificación actual (I, II, III, IV y NO APLICA)
o Informe de Anatomía Patológica
o Marcadores tumorales (en caso de corresponder)
o Estudios complementarios (en caso de corresponder)
- TAC
- PET
- RNM
- Centellograma
- Colonoscopia
Actualización
Semestral
1.
Resumen de Historia Clínica de seguimiento
2. Matrícula del Profesional Tratante
3. Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4. Información adicional
o ECOG (Eastern Cooperative Oncology
Group) de Seguimiento
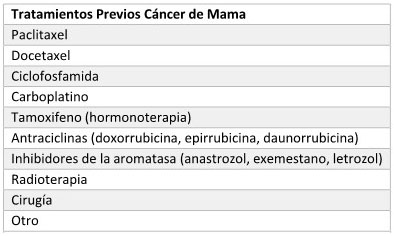
CÁNCER DE MAMA
Fundamento terapéutico
Cáncer de Mama HER2 Positivo
- Tratamiento en adyuvancia (hasta 12
meses)
- Tratamiento del cáncer avanzado
Cáncer de Mama Metastásico RE positivo, HER2 negativo
- Tratamiento del cáncer avanzado
Tratamientos Previos
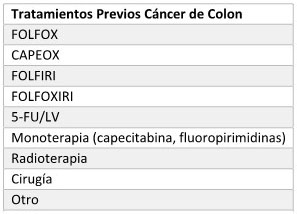
CÁNCER DE COLON
Fundamento terapéutico:
Tratamiento de pacientes con diagnóstico de carcinoma colorrectal (CCR)
avanzado o metastásico, en monoterapia o combinación con quimioterapia
citotóxica convencional clínicamente probada para CCR, con estado
funcional ECOG entre 0-1, mayores de18 años.
Tratamientos Previos
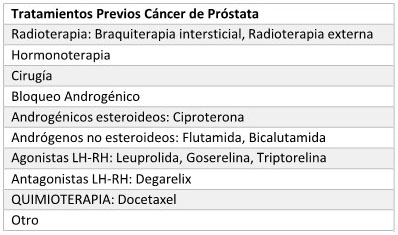
CÁNCER DE PRÓSTATA
Fundamento terapéutico
general:
Tratamiento de hombres adultos con cáncer de próstata metastásico
resistente a la castración cuya enfermedad ha progresado durante o tras
el tratamiento con docetaxel o no sean susceptibles de recibir
quimioterapia con docetaxel.
Tratamientos Previos
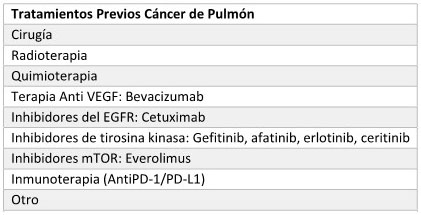
CÁNCER DE PULMÓN
CÁNCER DE PULMÓN NO MICROCÍTICO O NO
PEQUEÑAS CÉLULAS
Fundamento terapéutico
general:
Tratamiento de pacientes adultos con diagnóstico de cáncer de pulmón no
microcítico (CPNM) localmente avanzado o metastásico con estado
funcional ECOG 0 a 1.
Fundamento terapéutico
específico
- Inhibidores EGFR: presencia de la
mutación del factor de crecimiento epidérmico (EGFR) o mutación EGFR
T790m positiva según corresponda al producto
- Inhibidores ALK: presencia de la mutación de la quinasa del linfoma
anaplásico (ALK)
- Inmunoterapia: Sobreexpresión de ligando 1 de muerte programada PD-L1
(> 50%) - La utilización de inmunoterapia se limitará a dos años
Tratamientos Previos
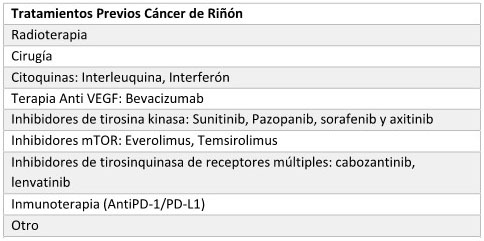
CÁNCER DE RIÑÓN
Fundamento terapéutico
general:
Tratamiento de pacientes adultos con diagnóstico decarcinoma de células
renales avanzado o metastásico con estado funcional ECOG entre 0 -1.
Tratamientos Previos
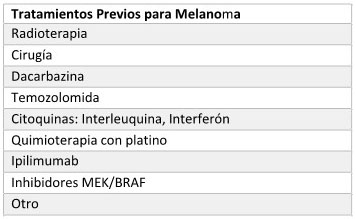
MELANOMA
Fundamento terapéutico
general: Tratamiento de pacientes mayores de 18 años con
diagnóstico de melanoma cutáneo avanzado, irresecable y/o metastásico.
Fundamento terapéutico
específico:
- Inhibidores BRAF: Mutación BRAF V600
positiva
- Inmunoterapia: PD-L1 > 1%
Tratamientos Previos
MÓDULO DE RADIOTERAPIA ONCOLÓGICA
Tecnologías incluidas
• Módulo de radioterapia tridimensional
conformada (RTC 3D)
• Radioterapia de intensidad modulada (IMRT)
La radioterapia conformacional de haz externo de alta energía es
aquella en la que se usa tecnología de avanzada para adaptar la
radioterapia a las estructuras anatómicas de cada paciente. Con la
ayuda de imágenes tridimensionales computarizadas, es posible moldear
el haz de radiación para que se ajuste a la forma de los tumores.
Actualmente hay dos niveles de radioterapia conformacional: la
radioterapia conformacional tridimensional (RTC 3D) y la radioterapia
deintensidad modulada (IMRT). Ambas modalidades permiten administrar
mayores dosis de radiación al tumor mientras se protegen los órganos
normales circundantes.
A. Módulo de radioterapia de
intensidad modulada (IMRT) para cáncer de próstata y cánceres de cabeza
y cuello
Los criterios para utilizar la IMRT en distintas localizaciones
anatómicas tumorales se fundamentan en mejorar la adaptación al volumen
tumoral con formas cóncavas, convexas o con invaginaciones y conseguir
dosis heterogéneas en el volumen tratado, adaptadas a la distinta
prescripción sobre zonas tumorales o ganglionares.
Fundamento terapéutico:
Cáncer de Próstata
■ Tumor localizado (ausencia de
extensión del tumor a ganglios linfáticos o metástasis a distancia) T1
N0 M0.
■ Buen estado general del paciente.
■ Edad menor a 65 años.
Cáncer de Cabeza y Cuello
■ Cáncer de cavidad oral y labios.
■ Cáncer de laringe, hipofaringe, orofaringe, nasofaringe.
■ Cáncer de senos paranasales y de cavidad nasal.
■ Cáncer de glándulas salivales.
■ Cáncer primario oculto en cabeza y región del cuello.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de inicio de radioterapia
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Protocolo de Anatomía Patológica
6. Prescripción de la IMRT
7. Protocolo de aplicación de IMRT
B. Módulo de radioterapia
tridimensional conformada (RTC 3D)
Fundamento terapéutico:
■ Cáncer de Cabeza y Cuello.
■ Tumores Cerebrales.
■ Cáncer de Laringe.
■ Cáncer de Esófago.
■ Cáncer de Mama.
■ Cáncer de Pulmón.
■ Cáncer de Páncreas.
■ Cáncer de Hígado.
■ Cáncer de Vejiga.
■ Cáncer de Recto.
■ Cáncer de Próstata.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de inicio de radioterapia
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Protocolo de Anatomía Patológica
6. Prescripción de la Radioterapia
Tridimensional Conformada
7. Protocolo de aplicación de
Radioterapia Tridimensional Conformada
ONCOHEMATOLOGÍA
MIELOMA MÚLTIPLE
Fundamento diagnóstico:
El diagnóstico de Mieloma Múltiple incluye las siguientes
características:
- Proteína monoclonal presente en suero
u orina
- Células Plasmáticas monoclonales > 10% en Médula Ósea (MO) o
biopsia de plasmocitoma
- Disfunción orgánica relacionada al Mieloma: a) Calcio elevado en
suero (>10.5 mg/dl); b) Insuficiencia Renal (Creatinina > 2
mg/dl); c) Anemia (Hb < 10 gr/dl o Hb < 2 gr de lonormal); d)
Lesión ósea lítica u osteoporosis.
El tratamiento actual para pacientes con mieloma múltiple sintomático
se puede dividir en inducción, de consolidación (que se utilizan menos
para los pacientes de edad muy avanzada), de mantenimiento y cuidados
médicos de soporte.
De acuerdo con la edad y características funcionales, los pacientes se
dividen en aquellos que son candidatos a trasplante de medula ósea y
aquellos que no.
Fundamento terapéutico:
- Tratamiento de primera línea en
esquemas de quimioterapia como inducción al trasplante en pacientes
candidatos, o en aquellos aptos para recibir quimioterapia no
candidatos a trasplante.
- Tratamiento de primera recaída y posteriores.
Los tratamientos para el mieloma múltiple deberán ser seleccionados de
acuerdo con las condiciones clínicas del paciente, las recomendaciones
de guías de práctica clínica, y el mejor perfil de
beneficios-riesgos-costos.
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del Profesional Tratante
4. Fecha de Inicio de Tratamiento
sujeto a recupero
5. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá
incluir Fundamento Terapéutico y los antecedentes de medicación
utilizada)
o Laboratorio
o Biopsia de Medula Ósea
o Citometría de flujo
Tratamientos Previos
Seguimiento
del Beneficiario:
Actualización Semestral
1-
Resumen de Historia Clínica de seguimiento
2- Actualización de la Información
Medica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
3- Matrícula Profesional Tratante
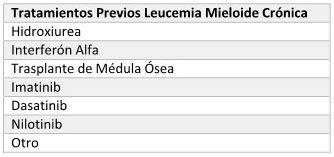
LEUCEMIA MIELOIDE CRÓNICA (LMC)
Fundamento Terapéutico:
Pacientes con diagnóstico de LMC. La elección del tratamiento se
realiza según diferentes variables como comorbilidades, escala de
riesgo (Sokal, Hasford, EUTOS, ELTS) y edad.
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Fase de LMC al diagnostico
o Crónica
o Acelerada
o Crisis Blástica
3. Tratamientos Previos
4. Matrícula del Profesional Tratante
5. Fecha de Inicio de Tratamiento
sujeto a recupero
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá
incluir Fundamento Terapéutico y los antecedentes de medicación
utilizada)
o Laboratorio
o Biopsia de Medula Ósea
o Citometría de flujo
o Determinación Cromosoma de Filadelfia y/o el gen BCR-ABL
o Mutación T3l5I (en caso de corresponder)
Tratamientos Previos
Seguimiento
del Beneficiario:
Actualización Semestral
1-
Resumen de Historia Clínica de seguimiento
2- Respuesta obtenida
o Respuesta hematológica (S-N)
o Respuesta citogenética (completa; parcial; menor; mínima; nula)
o Respuesta Molecular (mayor; menor; mínima; nula)
3- Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4- Matrícula Profesional Tratante
REUMATOLOGÍA
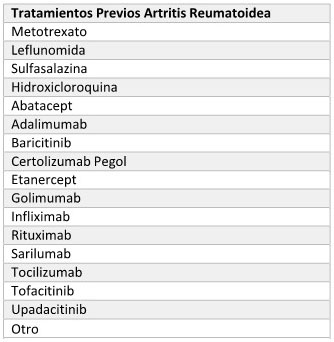
ARTRITIS REUMATOIDEA
Criterios de Diagnóstico
Presencia de al menos una articulación con sinovitis (inflamación) que
no se explique por otra causa y, adicionalmente, un puntaje de 6 o
mayor en ACR/EULAR 2010.
Estos criterios también permiten hacer el diagnóstico en aquellos
pacientes que presenten AR evolucionada siempre que: 1) Tengan
erosiones típicas de AR; 2) presenten una enfermedad de larga evolución
(activa o inactiva) cuyos datos retrospectivos permitan la
clasificación con los criterios mencionados.
Fundamento terapéutico:
Tratamiento de artritis reumatoidea activa moderada a grave, en
monoterapia o en combinacióncon metotrexato, que hayan presentado una
respuesta inadecuada o intolerancia a otros fármacos antirreumáticos
modificadores de la enfermedad (FAME), los cuales pueden incluir
también uno o más tratamientos con FAME biológicos o dirigidos.
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del Profesional Tratante
4. Fecha de Inicio de Tratamiento
sujeto a recupero
5. Score DAS 28 (Disease Activity
Score 28) de inicio
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá
incluir Fundamento Terapéutico y los antecedentes de medicación
utilizada)
Tratamientos Previos
Actualización
Semestral
1-
Resumen de Historia Clínica de seguimiento
2- Score DAS 28 (Disease Activity
Score 28) de seguimiento
3- Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4- Matrícula Profesional Tratante
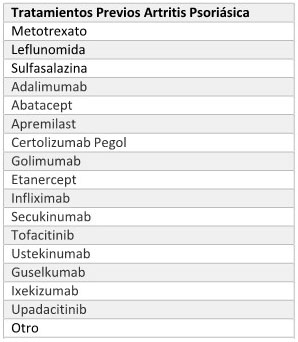
ARTRITIS PSORIÁSICA
Criterios de Diagnóstico
Los criterios diagnósticos más utilizados son los de CASPAR
(Classification criteria for Psoriatic Arthritis), que incluyen la
presencia de enfermedad inflamatoria articular (periférica, espinal o
entesítica) con 3 o más puntos en cualquiera de las 5 categorías
siguientes:
1) Psoriasis actual o historia personal o familiar de psoriasis.
2) Distrofia psoriásica ungueal.
3) Test negativo para el factor reumatoideo determinado por cualquier
método, excepto porlátex.
4) Historia actual de dactilitis.
5) Evidencia radiológica de neoformación ósea yuxtaarticular.
La presencia de psoriasis actual suma 2 puntos, el resto suma 1 punto.
Fundamento terapéutico:
En monoterapia o asociado a metotrexato en artritis psoriásica activa y
progresiva cuando la respuesta a la terapia previa con fármacos
antirreumáticos modificadores de la enfermedad (FAME) no ha sido
adecuada.
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del profesional Tratante
4. Fecha de Inicio de Tratamiento
sujeto a recupero
5. Score DAS 28 (Disease Activity
Score 28) de inicio
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá
incluir Fundamento Terapéutico y los antecedentes de medicación
utilizada)
o Biopsia de piel
Tratamientos Previos
Actualización
Semestral
1-
Resumen de Historia Clínica de seguimiento
2- Score DAS 28 (Disease Activity
Score 28) de seguimiento
3- Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4- Matrícula Profesional Tratante
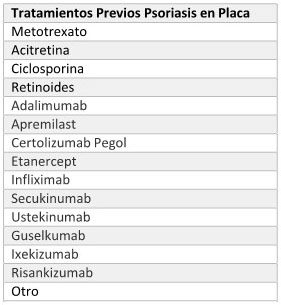
PSORIASIS EN PLACA
Criterios de Diagnóstico
El diagnóstico es clínico y se realiza con el clásico raspado metódico.
La realización de biopsiapara la confirmación histopatológica se deja
sólo para los casos de duda diagnóstica.
Fundamento terapéutico:
Pacientes adultos con psoriasis en placa moderada a severa, con
respuesta inadecuada o intolerancia a terapias sistémicas
convencionales (como por ejemplo metrotrexato o ciclosporina), FAMES
biológicos o dirigidos.
Información requerida:
Empadronamiento
del Beneficiario:
1. Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del Profesional Tratante
4. Fecha de Inicio de Tratamiento sujeto a recupero
5. Score PASI (Psoriasis Area Severity Index) de inicio
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá incluir Fundamento Terapéutico y
los antecedentes de medicación utilizada)
o Biopsia de piel
1- Resumen de Historia Clínica de
seguimiento
2- Score PASI (Psoriasis Area Severity Index) de seguimiento
3- Actualización de la Información Medica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4- Matrícula Profesional Tratante
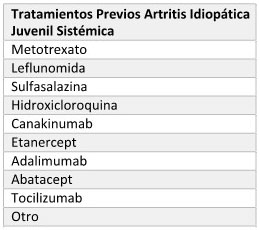
ARTRITIS IDIOPÁTICA JUVENIL SISTÉMICA
(AIJS)
Criterios de Diagnóstico
Criterios generales: a) artritis persistente de por lo menos 6 semanas
de duración en una o más articulaciones, de inicio antes de los 16
años; b) exclusión de otras causas de artritis.
Fundamento terapéutico:
Niños y adolescentes de entre 2 y 17 años que hayan presentado una
respuesta insuficiente a uno o más fármacos antirreumáticos
modificadores de la enfermedad (FAME) como monoterapia o combinado con
metrotexato
Información requerida:
Empadronamiento del Beneficiario:
1. Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del Profesional Tratante
4. Fecha de Inicio de Tratamiento sujeto a recupero
5. Score JADAS 27 (Juvenile Arthritis Disease Activity Score 27) de
inicio
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá incluir Fundamento Terapéutico y
los antecedentes de medicación utilizada)
o Biopsia de piel
Tratamientos Previos
Actualización
Semestral
1- Resumen de Historia Clínica de seguimiento
2- Score JADAS 27 (Juvenile Arthritis Disease Activity Score 27) de
seguimiento
3- Actualización de la Información Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4- Matrícula Profesional Tratante
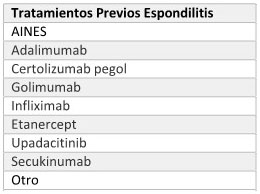
ESPONDILITIS ANQUILOSANTE-ESPONDILITIS
AXIAL NO RADIOGRÁFICA
Criterio diagnóstico:
Tres criterios clínicos o un criterio radiológico
sin otra causa que explique la patología. Criterios clínicos: a) dolor
lumbar mayor a 3 meses que mejora con el ejercicio y nocede con el
reposo; b) limitación de la movilidad de la columna lumbar en los
planos frontal y sagital; c) reducción de la expansión torácica
corregida por edad y sexo. Criterios radiológicos: a) sacroilitis grado
mayor a grado 2 bilateral; b) sacroilitis grado 3-4 unilateral.
La espondilitis axial no radiológica se considera un estadio previo a
la espondilitis anquilosante,para su diagnóstico no es necesario
demostrar sacroilitis radiológica.
Fundamento terapéutico:
Espondilitis anquilosante activa, con evidencia radiológica o sin ella,
en pacientes adultos que han respondido inadecuadamente al tratamiento
convencional.
Información requerida:
Empadronamiento
del Beneficiario:
1. Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del Profesional Tratante
4. Fecha de Inicio de Tratamiento sujeto a recupero
5. BASDAI (Bath Ankylosing Spondylitis Disease Activity Index) de inicio
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá incluir Fundamento Terapéutico y
los antecedentes de medicación utilizada)
o Estudios complementarios
para arribar al diagnóstico de laboratorio:
- HLA B27
- VSG (eritrosedimentación) o PCR (proteína C reactiva)
o Pruebas de Imágenes
- Radiografía simple de columna sacroilíaca
- Resonancia magnética sacroilíaca
1- Resumen de Historia Clínica de seguimiento
2- BASDAI de seguimiento
3- Actualización de la Información Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4- Matrícula Profesional Tratante
ENFERMEDAD INFLAMATORIA INTESTINAL
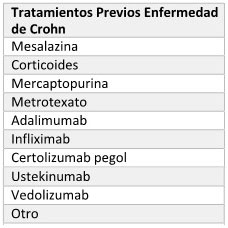
ENFERMEDAD DE CROHN
Criterios de Diagnóstico
Criterios generales: El diagnóstico de certeza de Enfermedad de Crohn
se realiza mediante la combinación de un criterio de biopsia (presencia
de granulomas o infiltrados linfoides), sumadoa la presencia de al
menos dos de los siguientes criterios: a) Lesión digestiva alta, b)
Lesión anal, c) Distribución segmentaría, d) Lesión transmural, e)
Fisura anal, f) Absceso, g) Fístula, i) Estenosis.
Fundamento terapéutico:
Pacientes adultos y niños, a partir de los 6 años, con enfermedad
activa moderada a severa que han tenido una respuesta inadecuada a la
terapia convencional.
Información requerida:
Empadronamiento
del Beneficiario:
1. Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del Profesional Tratante
4. Fecha de Inicio de Tratamiento sujeto a recupero
5. Crohn disease activity index (CDAI) de inicio
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá incluir Fundamento Terapéutico y
los antecedentes de medicación utilizada)
o Estudio de imagen
confirmatorio (Endoscopia-Biopsia-Enterotomografía- Enteroresonancia)
Tratamientos Previos
Actualización
Semestral
1- Resumen de Historia Clínica de
seguimiento
2- Crohn disease activity index (CDAI) de seguimiento
3- Actualización de la Información Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4- Matrícula Profesional Tratante
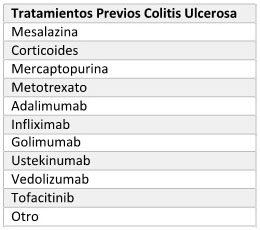
COLITIS ULCEROSA
Criterio diagnóstico:
La presentación más característica de la colitis ulcerosa es la diarrea
con sangre. Pueden asociarse otros síntomas como: dolor abdominal
(sobre todo en fosa ilíaca izquierda), fiebre en los brotes severos y
extensos, tenesmo rectal, urgencia o incontinencia en las formas de
afectación más distal, o síntomas de afectación general como pérdida de
peso y/o anorexia. Además, pueden aparecer manifestaciones
extraintestinales, fundamentalmente articulares, cutáneas y oculares.
Los hallazgos en la colonoscopía y anatomía patológica suelen ser
confirmatorios.
Fundamento terapéutico:
Colitis ulcerosa activa, moderada a grave, en pacientes que
respondieron inadecuadamente al tratamiento convencional, incluyendo
corticoesteroides y mecaptopurina o mesalazina, o que
presentanintolerancia o tienen contraindicaciones médicas al empleo de
estos.
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del Profesional Tratante
4. Fecha de Inicio de Tratamiento sujeto a recupero
5. Score de Mayo de inicio
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá incluir Fundamento Terapéutico y
los antecedentes de medicación utilizada)
o Estudio de imagen
confirmatorio (Endoscopia y/o Biopsia)
Actualización
Semestral
1- Resumen de Historia Clínica de seguimiento
2- Score de Mayo de seguimiento
3- Actualización de la Información Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4- Matrícula Profesional Tratante
ENFERMEDADES LISOSOMALES
ENFERMEDAD DE FABRY (EF)
Fundamento diagnóstico:
Varones (homocigotas): actividad disminuida de
la alfa galactosidasa en leucocitos. Mujeres (heterocigotas): actividad
disminuida de la alfa galactosidasa en leucocitos o confirmación
diagnóstica molecular (mutación para el gen galactosidasa alfa).
Fundamento terapéutico:
a) Pacientes con manifestaciones renales; b)
Pacientes conmanifestaciones severas no renales; c) Hombres
homocigotas, asintomáticos, con Enfermedad de Fabry clásica. En caso de
uso de Migalastat, los pacientes deberán ser portadores de mutaciones
susceptibles de respuesta al tratamiento.
Mujeres con Enfermedad de Fabry que presentan actividad enzimática poco
disminuida o normal deberán adjuntar el estudio genético que identifica
la mutacióncausante de EF en el gen GLA.
Información requerida:
Empadronamiento
del Beneficiario:
1. Fecha de Diagnóstico de la Patología
2. Matrícula del profesional Tratante
3. Fecha de Inicio de Tratamiento sujeto a recupero
4. Documentación Respaldatoria
o Resumen de Historia Clínica (debe detallar el cuadro clínico, y los
antecedentesde medicación utilizada)
o Laboratorio (Alfa Galactosidasa
A)
o Estudio genético gen GLA (en caso de corresponder)
o Mutaciones
susceptibles a Migalastat en caso decorresponder
5. Información Adicional
o Valor de Creatinina
o Valor de Proteinuria
o Tensión Arterial Sistólica
o Tensión Arterial Diastólica
Actualización
Semestral
1. Resumen de Historia Clínica de seguimiento
2. Actualización de la Información Medica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
3. Información adicional
o Valor de creatinina
o Valor de proteinuria
o Tensión Arterial Sistólica
o Tensión Arterial Diastólica
4. Matrícula del Profesional Tratante
ENFERMEDAD DE GAUCHER (EG)
Fundamento diagnóstico:
Disminución en la actividad de la enzima glucocerebrosidasa.
Fundamento terapéutico:
Terapia de sustitución enzimática a largo plazo en pacientes
pediátricos y adultos con diagnóstico confirmado de enfermedad de
Gaucher no neuropática (tipo 1) o neuropáticacrónica (tipo 3) que
presenten, además, manifestaciones no neurológicas clínicamente
importantes de la enfermedad.
Las manifestaciones no neurológicas de la enfermedad de Gaucher
incluyen una o más de las siguientes afecciones: 1) Anemia tras
exclusión de otras causas, tales como déficit de hierro; 2)
Trombocitopenia; 3) Enfermedad ósea tras exclusión de otras causas,
tales como déficit de Vitamina D; 4) Hepatomegalia o esplenomegalia.
Terapia de reducción de sustrato: pacientes adultos con enfermedad de
Gaucher tipo 1 únicamente en aquellos casos en los que no sea adecuado
el tratamiento enzimático sustitutivo. En al caso de Eliglustat solo
pacientes adultos con enfermedad de Gaucher tipo 1 que son
metabolizadores del CYP2D6 lentos, intermedios o rápidos.
Tratamientos incluidos:
- Terapia de sustitución enzimática (TSE)
- Terapia de reducción de sustrato (TRS)
Información requerida:
Empadronamiento
del Beneficiario:
1. Fecha de Diagnóstico de la Patología
2. Subtipo de E Gaucher (Tipo 1, Tipo 3)
3. Matrícula del Profesional Tratante
4. Tratamiento sujeto a recupero - (Terapia de Sustitución Enzimática /
Terapia de Reducción de Sustrato)
5. Tratamientos Previos
o Imiglucerasa
o Velaglucerasa Alfa
6. Fecha de Inicio de Tratamiento sujeto a recupero
7. Documentación Respaldatoria
o Resumen de Historia Clínica (debe detallar el cuadro clínico, y los
antecedentes de medicación utilizada)
o Laboratorio (Beta
Glucocerebrosidasa)
8. Información Adicional
o Hepatoesplenomegalia
o Hiperesplenismo
o Crisis Oseas
o Compromiso neurológico
Actualización
Semestral
1. Resumen de Historia Clínica de seguimiento
2. Actualización de la Información Medica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
3. Información adicional
o Hepatoesplenomegalia
o Hiperesplenismo
o Crisis Óseas
o Compromiso neurológico
4. Matrícula del Profesional Tratante
ENFERMEDAD DE POMPE
Fundamento diagnóstico:
Disminución en la actividad de la enzima alfa
glucosidasa ácida (AGA)en sangre, o por biopsia compatible.
Fundamento terapéutico:
Pacientes adultos y pediátricos con diagnóstico
confirmadode Enfermedad de Pompe, tanto en su variante temprana como
tardía.
Información requerida:
Empadronamiento
del Beneficiario:
1. Fecha de Diagnóstico de la Patología
2. Matrícula del Profesional Tratante
3. Fecha de Inicio de Tratamiento
sujeto a recupero
4. Documentación Respaldatoria
o Resumen de Historia Clínica (debe detallar el cuadro clínico, y los
antecedentes de medicación utilizada)
o Laboratorio/ Estudio Genético
(Alfa Glucosidasa Ácida)
o Biopsia (en caso de corresponder)
5. Información Adicional
o CVF (Capacidad vital forzada)
Actualización
Semestral
1. Resumen de Historia Clínica de seguimiento
2. Actualización de la Información Medica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
3. Información adicional
o CVF (Capacidad vital forzada)
4. Matrícula del Profesional Tratante
MUCOPOLISACARIDOSIS TIPO I
Fundamento diagnóstico:
Disminución en la actividad de la enzima alfa L-iduronidasa.
Fundamento Terapéutico:
a) Pacientes con Subtipo Hurler; b) Pacientes
con Subtipo Hurler-Scheie; c) Pacientes con Subtipo Scheie y síntomas
moderados a severos.
Información requerida:
Empadronamiento
del Beneficiario:
1. Fecha de Diagnóstico de la Patología
2. Subtipo de MPS I (Subtipo Hurler, Subtipo Hurler-Scheie,Subtipo
Scheie y síntomas moderados a severos)
3. Matrícula del Profesional Tratante
4. Fecha de Inicio de Tratamiento sujeto a recupero
5. Documentación Respaldatoria
o Resumen de Historia Clínica (debe detallar el cuadro clínico, y los
antecedentesde medicación utilizada)
o Laboratorio (Alfa L-Iduronidasa)
6. Información Adicional
o Compromiso cardíaco
o Compromiso pulmonar
o Compromiso auditivo
o Compromiso neurológico
Actualización
Semestral
1.
Resumen de Historia Clínica de seguimiento
2. Actualización de la Información
Medica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
3. Información adicional
o Compromiso cardíaco
o Compromiso pulmonar
o Compromiso auditivo
o Compromiso neurológico
4. Matrícula del Profesional Tratante
MUCOPOLISACARIDOSIS TIPO II
Fundamento diagnóstico:
Actividad disminuida de la enzima lduronato sulfatasa.
Fundamento terapéutico:
Pacientes con fenotipo leve. No se otorgará
tratamiento en lossiguientes casos: a) Formas graves o avanzadas, en
las que no se observa beneficios significativos; Pacientes con daño
neurológico o cognitivo severo.
Información requerida:
Empadronamiento
del Beneficiario:
7.
Fecha de Diagnóstico de la Patología
8. Matrícula del Profesional Tratante
9. Fecha de Inicio de Tratamiento
sujeto a recupero
10. Documentación Respaldatoria
o Resumen de Historia Clínica (debe detallar el cuadro clínico, y los
antecedentes de medicación utilizada)
o Laboratorio (lduronato
sulfatasa)
o Ecocardiograma
o Evaluación oftalmológica
Actualización
Semestral
1.
Resumen de Historia Clínica de seguimiento
2. Actualización de la Información
Medica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
3. Información adicional
o Ecocardiograma
o Evaluación oftalmológica
4. Matrícula del Profesional Tratante
MUCOPOLISACARIDOSIS TIPO VI
Fundamento diagnóstico:
Actividad disminuida de la enzima arilsulfatasa B (ASB).
Fundamento terapéutico:
Pacientes con diagnóstico confirmado de
Mucopolisacaridosis tipo VI. Se sugiere especialmente el tratamiento en
pacientes con daño de órgano blanco, los cuales deben cumplir al menos
uno de los siguientes criterios: a) apneas del sueño: más de 1
evento/hora en menores de 18 años o más de 5 eventos/hora en mayores de
18 años; b) saturación de oxígeno nocturna: < 92% en menores de 18
años o< 85% en mayores de 18 años,con capacidad vital forzada (CVF)
< 80% del predictivo para la edad; e) caminar menos de 350 men test
de caminata de 6 minutos; d) deterioro de función sistólica del
ventrículo izquierda (VI).
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Matrícula del Profesional Tratante
3. Fecha de Inicio de Tratamiento
sujeto a recupero
4. Criterios adicionales
5.
Documentación Respaldatoria
o Resumen de Historia Clínica (debe
detallar el cuadro clínico, y los
antecedentes de medicación utilizada)
o Laboratorio (Arilsulfatasa B)
Actualización
Semestral
1
.
Resumen de Historia Clínica de seguimiento
2. Actualización de la Información
Medica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
3. Matrícula del Profesional Tratante
:
Pacientes con diagnóstico confirmado de Atrofia
Muscular Espinal (AME), tipo I o II, sin comorbilidades que puedan
interferir con el proceso de administración de la tecnología. Los
pacientes deben ser evaluados por profesionales que acrediten
competencias en el manejo de la patología y un equipo
interdisciplinario. La decisión de iniciar tratamiento específico para
AME deberá justificar los potenciales beneficios a obtener del
tratamiento.
El recupero comprende a todo
tratamiento farmacológico destinado a
tratar la patología bajo los fundamentos terapéuticos descriptos. Queda
excluido el tratamiento sintomático (sostén nutricional, ventilatorio o
neuromuscular).
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Tipo de AME (I, II, III, IV)
3. Matrícula del profesional Tratante
4. Fecha de Inicio de Tratamiento
sujeto a recupero
5. Documentación Respaldatoria
o Resumen de Historia Clínica (debe
detallar el cuadro clínico, y los
antecedentes de medicación utilizada)
o Estudio Genético AME
o Dictamen favorable de CONAME (Comisión Nacional de Atrofia Muscular
Espinal del Ministerio de Salud) o manda Judicial de cobertura
6. Información Adicional
o Escala CHOP-INTEND Valor Basal
o HFMSE (Escala Motora Funcional de Hammersmith versión extendida)
Valor Basal
o Traqueotomía
o Deglución
o Requerimiento de apoyo respiratorio
o Cantidad de horas de apoyo respiratorio
Actualización
Semestral
1.
Resumen de Historia Clínica de seguimiento
2. Matrícula del Profesional Tratante
3. Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4. Información adicional
o Escala CHOP-INTEND
o Escala HINE-2 (Hammersmith Infant Neurological Examination Section 2)
Valor Seguimiento
o HFMSE (Escala Motora Funcional de Hammersmith versión extendida)
Valor Seguimiento
o Traqueotomía
o Deglución
o Requerimiento de apoyo respiratorio
o Cantidad de horas de apoyo respiratorio
TIROSINEMIA TIPO I
Fundamento diagnóstico:
1) Exámenes bioquímicos: a) Niveles plasmáticos
elevados de tirosina (> 200 ^mol/l), metionina y de fenilalanina, b)
Hiperaminoaciduria generalizada, c) Niveles aumentados en orina de los
ácidos 4-hidroxifenilderivados, Niveles aumentados de succinilacetona
en plasma, d) Medida de la actividad PBG-S o d-ALAD en sangre total
heparinizada e) Medida de la actividad FAH en linfocitos, fibroblastos
de piel cultivados, biopsia hepática y/o eritrocitos, que se encuentra
muy disminuida. 2) Estudios genéticos, sobre los cuales pueden
presentarse una gran variabilidad de mutaciones.
Fundamento terapéutico:
Tratamiento de pacientes con diagnóstico
confirmado de tirosinemia hereditaria tipo I (TH-1) en combinación con
dieta restrictiva de tirosina y fenilalanina.
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Matrícula del Profesional Tratante
3. Fecha de Inicio de Tratamiento
sujeto a recupero
4. Documentación Respaldatoria
o Resumen de Historia Clínica (debe
detallar el cuadro clínico y el tratamiento dietario)
o Laboratorio (aminoácidos y ácidos orgánicos)
o Estudio Genético mutaciones gen FHA
Actualización
Semestral
1.
Resumen de Historia Clínica de seguimiento
2. Matrícula del Profesional Tratante
3. Actualización de la Información
Medica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
HEMOGLOBINURIA PAROXÍSTICA NOCTURNA
Fundamento terapéutico:
Pacientes mayores de 18 años, con diagnóstico
de Hemoglobinuria Paroxística Nocturna, con antecedentes de por lo
menos cuatro transfusiones en los últimos 12 meses, que no se
encuentren cursando infección aguda por Neisseria meningitidis, y
cuenten con vacunación para Neisseria meningitidis al menos 2 semanas
previas al inicio del tratamiento.
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Matrícula del Profesional Tratante
3. Fecha de Inicio de Tratamiento
sujeto a recupero
4. Documentación Respaldatoria
o Resumen de Historia Clínica (detalle
de antecedentes, estadio
evolutivo y tratamiento utilizados)
o Laboratorio HPN (Plaquetas,
Hemoglobina, Creatinina)
o Citometría de Flujo
Actualización
Semestral
1. Resumen de Historia Clínica de
seguimiento
2. Matrícula del Profesional Tratante
3. Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4. Información adicional
o Laboratorio HPN (Plaquetas,
Hemoglobina, Creatinina)
SÍNDROME URÉMICO HEMOLÍTICO ATÍPICO
Fundamento diagnóstico:
El SUHa se considera un "diagnóstico de
exclusión", con la presentación clínica habitual a los síndromes de
microangiopatía trombótica que comprende una tríada compuesta por
trombocitopenia, anemia hemolítica de carácter microangiopático y daño
de órgano blanco, como se refirió habitualmente renal, y con alguna
frecuencia algún otro.
En este sentido, el cuadro clínico comprende:
• Trombocitopenia menor a 150.000/mm3 o
disminución mayor al 25% respecto de basal.
• Anemia hemolítica microangiopática (hemoglobina disminuida con LDH
elevada ypresencia de esquistocitos frecuentemente en el frotis
sanguíneo)
• Insuficiencia renal (creatinina sérica elevada y/o índice de filtrado
glomerular estimado descendido), o compromiso demostrable en otro
órgano blanco
Exámenes complementarios
solicitados para descartar otras causas de MAT:
• Determinación de infección por E.
Coli O157:H7 (Aislamiento por cultivo, FilArray y STx 1 y 2 u otros
ensayos que verifiquen presencia de E. coli enterotóxica).
• Determinación actividad enzimática ADAMTS13 mayor al 5-10%.
Otros exámenes
complementarios que pueden colaborar en el diagnóstico de SUHa:
• Actividad del complemento: C3, C4,
CH50.
• Biopsia de riñón con demostración de MAT histológica.
• Test genético que demuestre anomalías en los genes que regulan la vía
alterna del complemento.
Fundamento terapéutico:
Pacientes adultos y pediátricos con diagnóstico
de SHU atípico, con manifestaciones microangiopáticas, que no se
encuentren cursando infección aguda por Neisseria meningitidis, y
cuenten con vacunación para Neisseria meningitidis al menos 2 semanas
previas al inicio del tratamiento.
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Matrícula del Profesional Tratante
3. Manifestaciones Clínicas (IRA,
Anemia Hemolítica microangiopática,
Trombocitopenia grave, microangiopatía selectiva a nivel renal)
4. Manifestaciones Isquémicas otros
órganos blancos (SNC, Páncreas, Miocardio)
5. Fecha de Inicio de Tratamiento
sujeto a recupero
6. Documentación Respaldatoria
o Resumen de Historia Clínica (detalle
de antecedentes, estadio evolutivo y tratamiento utilizados)
o Laboratorio SUHa (Plaquetas, Hemoglobina, Creatinina)
Actualización
Semestral
1.
Resumen de Historia Clínica de seguimiento
2. Matrícula del Profesional Tratante
3. Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
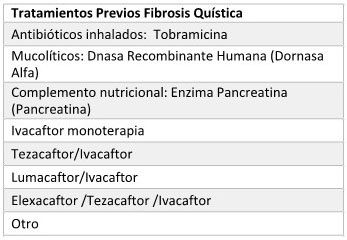
ENFERMEDAD FIBROQUÍSTICA DEL PÁNCREAS
Se encuentran comprendidos en recupero los pacientes con diagnóstico
establecido de Enfermedad Fibroquística del Páncreas.
Fundamento diagnóstico:
1) Test del sudor anormal (Cloro > 60 mEq/L), con una segunda prueba
que lo confirme o
2) Estudio molecular que documente la presencia de mutaciones del
Factor Regulador de la Conductancia Transmembrana (CFTR) o
3) Demostración de diferencia de potencial nasal transepitelial anormal.
Existen
casos atípicos en los cuales el test de sudor puede estar
dentro de los límites normales o con valores limítrofes y se debe
confirmar el diagnóstico sólo con el estudio molecular. En este último
caso deben estar presente 2 mutaciones para arribar al diagnóstico.
Fundamento terapéutico
Pacientes con diagnóstico confirmado de Fibrosis Quística, que se
adecuen el tratamiento a su condición clínica y estado mutacional.
Módulo 1: Tratamiento Sintomático Convencional de la FQ
Módulo 2: Tratamiento que incluye Terapias Moduladoras del CFTR
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del Profesional Tratante
4. Fecha de Inicio de Tratamiento
sujeto a recupero
5. Módulo sujeto a recupero (Modulo 1
- Modulo 2)
6. Clase de Mutación (2, 3, 4, 5, 6,
NA)
7. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá
incluir Fundamento Terapéutico y antecedentes de medicación utilizada).
o Test de Sudor
o Estudio Molecular donde se documentan al menos 2 mutaciones del CFTR
(en caso de solicitar recupero Modulo 2)
o Diferencia del Potencial
nasal transepitelial (solo en caso de corresponder)
8. Información Adicional
o Peso
o Talla
o VEF1 (volumen espiratorio forzado en 1 segundo)
o CVF (capacidad vital forzada)
Actualización
Semestral
1.
Resumen de Historia Clínica de seguimiento
2. Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
3. Información Adicional
o Peso
o Talla
o VEF1 (volumen espiratorio forzado en 1 segundo)
o CVF (capacidad vital forzada)
4. Matrícula Profesional Tratante
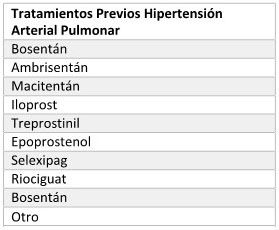
HIPERTENSIÓN ARTERIAL PULMONAR (HAP)
Fundamento diagnóstico:
Es obligatoria la realización de cateterismo
cardíaco derecho (CCD). Se define como HTP a la elevación de la presión
media de la arteria pulmonar (PMAP) > 25 mmHg, medida mediante CCD
con el paciente en reposo. Para el diagnóstico de HAP se requiereademás
la presencia de una presión de enclavamiento pulmonar (Wedge) (PAWP)
< 15 mmHg y resistencias vasculares pulmonares (RVP) > 3 unidades
Wood (UW) en ausencia de otras condiciones de HTP pre- capilar, como la
que puede darse en forma secundaria a enfermedadespulmonares, HPETC.
Fundamento terapéutico:
Pacientes con Hipertensión Arterial Pulmonar (grupo 1) con clase
funcional grado II - IV de la clasificación de la Organización Mundial
de la Salud (OMS).
Pacientes con Hipertensión Pulmonar Secundaria a Enfermedad
Tromboembólica Crónica (grupo 4), con contraindicación debidamente
justificada para la endarterectomía pulmonar o con hipertensión
pulmonar persistente o recurrente luego de la endarterectomía pulmonar,
conclase Funcional II a III de la OMS.
Grupos incluidos
- Grupo 1
- Grupo 4
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del profesional Tratante
4. Fecha de Inicio de Tratamiento
sujeto a recupero
5. Clase Funcional NYHA de inicio
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá
incluir Fundamento Terapéutico y
los antecedentes de medicación utilizada).
o Clasificación Etiológica
de la HAP (Grupo 1 - Grupo 4)
o Cateterismo de cavidades derechas
o Estudio que demuestre Tromboembolismo Pulmonar (solo en caso de grupo
4)
Tratamientos Previos
Actualización
Semestral
1.
Resumen de Historia Clínica de seguimiento
2. Clase Funcional NYHA de seguimiento
3. Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4. Matrícula Profesional Tratante
ESCLEROSIS MÚLTIPLE
Se encuentran comprendidos en recupero los pacientes con diagnóstico
establecido deEsclerosis Múltiple.
Fundamento diagnóstico:
Se diagnostica mediante los criterios de McDonald modificados (2017):
a. Al menos dos ataques clínicos; evidencia clínica objetiva de al
menos dos lesiones o evidenciaclínica de una lesión con constatación de
Historia Clínica razonable de un ataque previo. No se necesitan datos
adicionales para el diagnóstico.
b. Al menos dos ataques clínicos; evidencia clínica objetiva de una
lesión. Se necesita demostrar diseminación en espacio (DIS), por al
menos una lesión sintomática o asintomática típica de EMen T2 como
mínimo en dos áreas típicas del SNC: periventricular, yuxtacortical,
médula espinal o infratentorial o esperar un ataque clínico adicional
en un sitio diferente del SNC.
c. Un ataque clínico: evidencia clínica objetiva de al menos dos
lesiones. Se necesita demostrar uno de estos criterios: Diseminación en
tiempo (DIT) mediante la presencia de lesiones sintomáticas o
asintomáticas gadolinio (Gd) positivas y no Gd positivos simultáneas o
nueva lesión en T2 ó Gd positiva en el seguimiento por RMN, en
comparación con la RMN basal (independientemente del tiempo
transcurrido del estudio basal), o espera de un segundo ataque clínico;
o Bandas Oligoclonales en LCR (negativas en suero).
d. Un ataque clínico: evidencia clínica objetiva de una lesión
(síndrome clínico aislado). Se necesita demostrar DIT y DIS descritos
anteriormente.
e. Progresión neurológica insidiosa sugestiva de EM.
También será criterio diagnóstico, demostrar un año de progresión de la
discapacidad (prospectivo o retrospectivo) y dos de estos criterios: al
menos una lesión, sintomática o asintomática típica de EM en T2:
periventricular, yuxtacortical, médula espinal o infratentorial o dos o
más lesiones en médula espinal o bandas oligoclonales en LCR (negativas
en suero).
En el año 2017 se han revisado los criterios y la presencia de un
síndrome clínicamente aislado típico y una demostración clínica o de RM
de diseminación en el espacio, la presencia de bandas oligoclonales
específicas de LCR y negativas en el suero permite un diagnóstico de EM.
Fundamento terapéutico:
Tratamiento de pacientes adultos con esclerosis múltiple
remitente-recurrente y esclerosis múltiple progresiva primaria.
Las tecnologías listadas previamente pueden ser utilizadas de manera
individual como tratamiento modificador de la enfermedad en pacientes
con EM. La posología de estos debe ser realizada conforme lo
establecido en los respectivos prospectos aprobados por la ANMAT.
Información requerida:
Empadronamiento
del Beneficiario:
1.
Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Matrícula del Profesional Tratante
4. Valor de EDSS (Expanded Disability
Status Scale ) de inicio
5. Fecha de Inicio de Tratamiento
sujeto a recupero
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá
incluir Fundamento Terapéutico y
Diagnóstico según criterios de McDonald).
o Estado Evolutivo/Forma
Clínica (EMPP - EMRR)
o Resonancia magnética Nuclear
o Punción Lumbar / Análisis de líquido cefalorraquídeo (si
correspondiera)
Tratamientos Previos
Actualización
Semestral
1.
Resumen de Historia Clínica de seguimiento
2. EDSS (Expanded Disability Status
Scale) de seguimiento
3. Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4. Matrícula Profesional Tratante
DÉFICIT DE HORMONA DE CRECIMIENTO
Fundamento diagnóstico:
Niveles inferiores a los normales de Hormona de
Crecimiento, que ocasionen repercusión clínica, de etiología congénita
o adquirida.
Fundamento terapéutico:
Pacientes con talla inferior al percentil 3 ajustado a la edad y alguna
de las siguientes condiciones
• Síndrome de Prader Willi
• Déficit de Hormona de Crecimiento
• Síndrome de Turner
• Insuficiencia renal Crónica en la infancia
• Retardo del Crecimiento Intrauterino
No se reconocerá la cobertura del apoyo financiero solicitado en los
siguientes casos:
• Niños con baja estatura idiopática.
• Niños que están recibiendo Hormona de Crecimiento y que presenten:
o
Edad ósea igual o mayor a 14 años en niñas y 16 años en varones.
o Incremento de velocidad de crecimiento menor a 2 cm. por año luego de
un año de tratamiento.
o Cierre de los cartílagos de crecimiento.
1.
Fecha de Diagnóstico de la Patología
2. Patología asociada (Síndrome
de Prader Willi, Déficit de Hormona de Crecimiento, Síndrome de Turner,
Insuficiencia Renal crónica en la Infancia, Retardo del Crecimiento
Intrauterino)
3. Matrícula del Profesional Tratante
4. Fecha de Inicio de Tratamiento
sujeto a recupero
5. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá
incluir Fundamento Terapéutico y
los antecedentes de medicación utilizada).
o Nivel sérico de Hormona de
Crecimiento
o Estudio Genético
o Tabla de crecimiento pondoestatural
o
Edad ósea
6. Información adicional
o Talla o Peso
Actualización
Semestral
1.
Resumen de Historia Clínica de seguimiento
2. Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
3. Documentación Respaldatoria
o Edad ósea
o Velocidad de crecimiento
4. Información adicional
o Talla
o Peso
5. Matrícula Profesional Tratante
TRASPLANTE DE ÓRGANOS Y PRECURSORES
HEMATOPOYÉTICOS
NORMAS GENERALES DE TRASPLANTE
Los prestadores, sean públicos o privados, deberán estar habilitados
(tanto la Institución como el Equipo de Trasplante) por el INSTITUTO
NACIONAL CENTRAL UNICO COORDINADOR DE ABLACION E IMPLANTE (INCUCAI).
El INCUCAI gestionará los reintegros ante la Gerencia de Control
Prestacional a través de los mecanismos establecidos en el marco del
convenio que aprueba la Resolución N° 1627/2014- S.S.SALUD.
Módulo Pre-trasplante
El módulo comprende la realización de los estudios necesarios para
decidir un trasplante. Se deberán efectuar todos los exámenes
complementarios racionales y necesarios, así como también las
interconsultas especializadas (y los estudios y/o prácticas de ellas
derivadas), adecuados para una correcta evaluación de la condición de
receptor. En caso de donante vivo relacionado se deberán efectuar todos
los estudios necesarios en el candidato a donar, para unacorrecta
evaluación de su condición de donante. Deberá incluirse el informe
psico-social producido por el Centro de Trasplante, a fin de evaluar el
contexto socioambiental para garantizar las condiciones para una mejor
evolución del trasplante. En caso de donante vivo relacionado se
reintegrará únicamente el valor del estudio pre-trasplante del donante
que ha sido seleccionado como dador.
Para poder acceder al reintegro del Módulo Pre-trasplante se deberá
demostrar fehacientemente que el tiempo que medie entre el diagnóstico
de insuficiencia orgánica con indicación de trasplante y la
finalización del estudio que defina la condición de candidato o no al
mismo, no deberá superar los tres (3) meses para los trasplantes de
órgano único, salvo riñón,que será de seis (6) meses. Para los
trasplantes combinados este período será de seis (6) meses.
Para el caso específico de trasplante de riñón o reno páncreas este
período se contará desde lafecha Alta en el Registro Nacional de
Insuficiencia Renal Crónica Terminal del INCUCAI o bien desde el
diagnóstico e indicación de trasplante para aquellos casos en los que
el paciente no ingresa nunca en hemodiálisis.
En los casos en que se hubieran efectuado prácticas de pre-trasplante y
se hubiese producido el fallecimiento del paciente beneficiario, se
admitirá la solicitud de reintegro para la cobertura financiera de
dichas prácticas, debiéndose presentar el certificado de defunción y la
constanciade la realización de estas
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de la inscripción en el
Módulo Pre-trasplante
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el Trasplante
5. Resumen de estudios realizados como
Pre-tasplante
Módulo de Trasplante de órgano
Este módulo abarca todos los trasplantes de órganos listados en la
presente normativa.El módulo trasplante incluye:
• Ablación del órgano: en todos los
órganos salvo en riñón que la
ablación la efectúa el INCUCAI. En los restantes órganos sólidos la
ablación está a cargo del Equipo detrasplante.
• Internación en los diferentes
sectores acorde a necesidad.
• Honorarios del equipo médico clínico, quirúrgico, de anestesia,
interconsultores y técnicos intervinientes en la cirugía y durante la
internación.
• Gastos quirúrgicos, derechos
quirúrgicos, de anestesia.
• Estudios complementarios necesarios relacionados con el
procedimiento, de cualquiernivel de complejidad, que requieran ser
efectuados estando el paciente internado.
• Medicamentos y material descartable
utilizados durante la cirugía y en la internación.
• Medicina transfusional.
• Estudios endoscópicos necesarios.
• Estudios hemodinámicos.
• Estudios de Anatomía Patológica (biopsias de todo tipo).
• Todas las reoperaciones por complicaciones propias de la cirugía
original.
1.
Fecha de inscripción en registro del INCUCAI
2. Fecha del procedimiento
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el trasplante
5. Protocolo de Trasplante o
Certificado de Trasplante del INCUCAI
Módulo de Trasplante de
Precursores Hematopoyéticos
Este módulo es aplicable indistintamente a las tres variedades de
trasplante:
- Autólogo
- Alogénico con donante emparentado
- Alogénico con donante no emparentado.
Difieren por la fuente de obtención de las células precursoras
hematopoyéticas.
Bajo este nombre se engloba al Trasplante de Precursores
Hematopoyéticos (Células Madre)- Lafuente de origen y extracción de las
células madre puede ser la médula ósea o bien la sangre periférica. La
sangre del cordón umbilical que se encuentra en el cordón y la placenta
después del parto es otra fuente de células madre progenitoras
utilizadas en la práctica clínica en el ámbito de los trasplantes
alogénicos.
Si las células proceden del propio paciente, se denomina trasplante
autólogo, si provienen de un donante celular distinto del paciente se
denomina trasplante alogénico.
El trasplante alogénico tiene a su vez distintas variedades según el
donante y la similitud del sistema de Antígenos Leucocitarios Humanos
(Human Luekocyte Antigens - HLA). Si el donante es un hermano gemelo
univitelino se denomina trasplante singénico. Cuando el donante es un
familiar HLA idéntico (en general, un hermano), se denomina trasplante
alogénico de hermano HLA idéntico. En el caso de que el donante sea un
familiar que comparte un solo haplotipo del sistema HLA se denomina
trasplante haploidéntico, y el donante puede ser un familiar cualquiera
(padre, madre, primos) que comparte sólo la mitad de los genes
implicados en el sistema HLA.
En el caso de un donante no emparentado se denomina trasplante de
donante no emparentadoy en este caso se procede a la activación de la
Búsqueda Internacional y a la Procuración de un donante a través del
INCUCAI.
El procedimiento del trasplante consta de diferentes etapas sucesivas a
saber: selección del dador, administración de agentes de movilización
celular, extracción y aféresis, preparación del producto obtenido para
su conservación, criopreservación, administración del régimen
preparatorio al paciente, trasplante por infusión de las células madre
obtenidas, arraigo del injerto y recuperación.
Fundamento Terapéutico: Según las indicaciones validadas por el INCUCAI
(Resolución 309/07 INCUCAI y su Modificatoria 414/12).
La composición del Módulo Incluye:
• Honorarios médicos de la totalidad
del equipo de trasplante,
incluyendo todas las especialidades médicas que se requieran durante la
internación.
• Internación en habitación individual con aire filtrado y climatizada,
con baño privado, en la Unidad de Trasplante de Médula Ósea. Medidas de
aislamiento. Personal de enfermería especializado para el cuidado de
pacientes neutropénicos y trasplantados. Alimentación balanceada y
descontaminada bajo supervisión de Nutricionista. Eventual alimentación
parenteral.
• Derechos asistenciales y de quirófano, colocación de catéteres,
prácticas y estudios necesarios inherentes a la patología de base
durante el procedimiento (análisis de laboratorio clínico,
hematológicos, bacteriología y virología).
• Anatomía Patológica, Inmunología.
• Diagnósticos por Imágenes: Radiología, Ecografía, TAC y RMN.
• Hemoterapia: Procesamiento y Transfusión de Glóbulos rojos, Plaquetas
y Hemoderivados, irradiación de los Hemoderivados.
• Medicamentos y material descartable: Citostáticos e Inmunosupresores,
Antibióticos, Soluciones parenterales, antifúnguicos y antivirales,
Kits para separación celular, Set de infusión para Alimentación
parenteral y enteral y todo otro material necesario para los
procedimientos.
• Implementación de técnicas específicas para la recolección de células
progenitoras del propio paciente (trasplante autólogo) o de dador
relacionado (trasplante alogénico con donante relacionado)
• Aféresis celular.
• Recuentos celulares del producto obtenido por citometría de flujo
para determinar el número de células progenitoras (CD34) Procesamiento
de las células previo a la preservación. Criopreservación y
conservación de Nitrógeno líquido.
• Drogas y factores estimulantes que se requieran para el procedimiento
de movilizaciónde células progenitoras de sangre periférica.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de realización del trasplante
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Protocolo de la infusión celular
6. Constancia del Registro de
Trasplantes de Precursores Hematopoyéticos del INCUCAI
7. Comprobante de colocación del CDI
8. Protocolo del procedimiento del CDI
INMUNOSUPRESIÓN POST TRASPLANTE
Fundamento terapéutico:
Pacientes con trasplante de órgano sólido que requieren tratamientocon
inmunosupresión
Fundamentos terapéuticos
específicos
Drogas inmunodepresoras en la etapa previa, posterior o concomitante a
la realización de los trasplantes, de por vida del paciente. Para
cualquier órgano o tejido, y como agente único o asociado a
corticoides. El uso deberá ajustarse a lo normado por la ANMAT y lo
recomendado en Guías de Práctica Clínica.
Información requerida
Empadronamiento
del Beneficiario:
1.
Fecha de trasplante
2. Órgano trasplantado
3. Tratamientos Previos
4. Matrícula del Profesional Tratante
5. Fecha de Inicio de Tratamiento
sujeto a recupero
6. Documentación Respaldatoria
o Resumen de Historia Clínica (deberá incluir Fundamento Terapéutico y
los antecedentes de medicación utilizada).
o Protocolo de trasplante o
certificado de trasplante del INCUCAI
o Serología para virus Epstein
Barr (en caso de corresponder)
Seguimiento
del Beneficiario:
Actualización Semestral
1.
Resumen de Historia Clínica de seguimiento
2. Actualización de la Información
Médica
a. Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
3. Matrícula del Profesional Tratante
INFECCIÓN POR VIRUS DE LA
INMUNODEFICIENCIA HUMANA (HIV)
Fundamento diagnóstico
Paciente adulto o niño mayor a 18 meses en quien se determine al menos
una prueba de tamizaje (por metodología ELISA, aglutinación de
partículas o test rápido) positiva para VIH con un test confirmatorio
por carga viral. En niños menores de 18 meses se considera el
diagnóstico con dos pruebas de PCR positivas para VIH.
Fundamento terapéutico:
Pacientes con infección por VIH, independientemente del recuento de
células CD4.
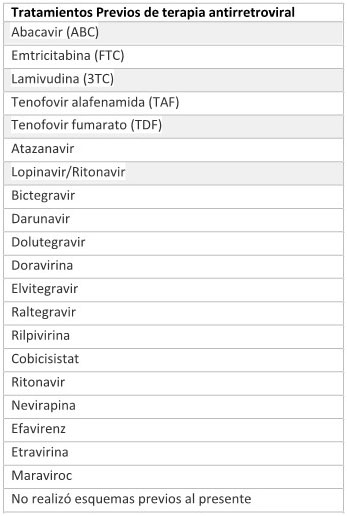
Drogas incluidas
Sub-Módulo A
Drogas base del esquema
- Abacavir (ABC)
- Emtricitabina (FTC)
- Lamivudina (3TC)
- Tenofovir alafenamida (TAF)
- Tenofovir fumarato (TDF)
Sub-Módulo B.1
Drogas base del esquema
- Atazanavir
- Lopinavir/Ritonavir
- Bictegravir
- Darunavir
- Dolutegravir
- Doravirina
- Elvitegravir
- Raltegravir
- Rilpivirina
- Cobicisistat *
- Ritonavir *
*Para asociar con Inhibidores de la Proteasa
Sub-Módulo B.2
Drogas base del esquema
- Efavirenz
- Nevirapina
Todo esquema de tratamiento debe incluir drogas del módulo A asociadas
a las del módulo B. Existe situaciones específicas donde se podrán
utilizar otras combinaciones recomendadas en Guías del Ministerio de
Salud o Sociedades Científicas, las cuales serán validadas por la
Gerencia de Gestión Estratégica.
Módulo C: Multifallo
Para el caso de esquemas de multifallo, se reconocerán las siguientes
drogas, que deberán reintegrarse adicionalmente a los módulos
previamente descriptos (A+B.1) o (A + B.2) u otros específicamente
validados.
- Dolutegravir (doble dosis)
- Etravirina (ETV)
- Maraviroc (MRV)
La Terapia Antirretroviral (TARV) a utilizar debe seguir las
recomendaciones vigentes de las guíasde práctica clínica y adecuarse a
los fines de garantizarla adherencia.
Fallo de tratamiento: se define como fallo virológico a la presencia de
carga viral (CV) plasmáticapor encima del límite de detección después
de al menos 24 semanas de tratamiento. Este dato debe ser confirmado en
2 muestras consecutivas. Si la carga viral plasmática es muy elevada
antes del inicio del TARV puede requerirse más de 24 semanas para
lograr la no detectabilidad.También constituye fallo virológico cuando
la CV aumenta por encima del límite de detección luego de haber
alcanzado la no detectabilidad, dato que requiere también confirmación
con unasegunda muestra. Se considera fallo al valor de CV mayor a 200
copias/ml.
El esquema utilizado después del primer fallo y subsiguientes,
dependerá del test de resistenciay de la historia de drogas
antirretrovirales utilizadas. Se considerará como fallo virológico a la
situación clínica donde la CV y la Historia Clínica lo confirmen, aun
sin la disponibilidad del test de resistencia, cuando dicha situación
sea justificada por causales válidas. La condición de multifallo solo
podría ser considerada a partir de un segundo fallo.
El cambio de TARV en pacientes con CV suprimida (no detectable), puede
ser realizado luego de objetivar al menos 6 meses de carga viral no
detectable. Puede responder a:
• Simplificación: utilización de un
esquema de menos comprimidos o menor dosis
• Intolerancia o toxicidad: Se modifica la droga a la que se atribuye
la intolerancia o toxicidad y se la reemplaza.
• Interacciones medicamentosas
• Embarazo
En todos estos casos, en los cuales el cambio se realiza con carga
viral suprimida, no se requiere test de resistencia.
Información Requerida
Empadronamiento
del Beneficiario:
1.
Resumen de Historia Clínica
2. Fecha de diagnóstico
3. Tratamientos Previos
4. Matrícula del Profesional Tratante
5. Fecha de Inicio de Tratamiento
sujeto a recupero
6. Constancia de denuncia al Programa
de Vigilancia del Ministerio de Salud de la Nación
7. Información Adicional
o Ultima Carga Viral
o Ultimo valor de CD4 en valores absolutos
o Ultimo valor de CD4 en valores porcentuales
Seguimiento
del Beneficiario:
Actualización Anual
1.
Resumen de Historia Clínica de seguimiento
2. Matrícula del Profesional Tratante
3. Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
4. Información Adicional
o Ultima Carga Viral
o Ultimo valor de CD4 en valores absolutos o Ultimo valor de CD4 en
valores porcentuales
HEPATITIS CRÓNICA POR VIRUS C
Fundamento terapéutico:
Todos los pacientes con diagnóstico de
infección por virus de Hepatitis C, independientemente de la gravedad
de la afección.
Todas las personas portadoras de VHC confirmado por ARN serán
candidatos a tratamiento antiviral con nuevos antivirales de acción
directa, independientemente de la gravedad de la fibrosis hepática.
Solo se podrán presentar ante SUR los
esquemas finalizados, en
los cuales se haya podido comprobar la respuesta viral obtenida.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Tratamientos Previos
3. Fecha de inicio de terapia
antiviral con esquema libre de interferón
4. Matrícula de Profesional Tratante
5. Resumen de Historia Clínica que
justifique el uso de la tecnología
6. Información clínica para aportar
o Genotipo viral
o Estadio de fibrosis
o Coinfección con VIH
o Antecedentes de trasplante de hígado
o Carga viral inicial
o Cantidad de semanas de tratamiento
o Respuesta viral obtenida
ANEURISMA DE AORTA ABDOMINAL /
ANEURISMA DE AORTA TORÁCICA
Tecnología:
Endoprótesis para tratamiento endovascular del aneurisma de aorta
abdominal o aneurisma de aorta torácica.
Fundamento terapéutico:
Se reconocerá el reintegro cuando se encuentre
justificada la imposibilidad de realizar la cirugía a cielo abierto y
cumpla con alguno de los criterios predictoresde riesgo:
- Infarto de miocardio agudo o reciente
con evidencia de riesgo isquémico determinado por síntomas y/o estudios
no invasivos.
- Angina inestable (CF III o IV).
- Arritmias significativas: bloqueo AV de alto grado / arritmias
ventriculares sintomáticas / arritmias supraventriculares con ritmo
ventricular no controlado.
- Enfermedad valvular severa.
- Enfermedad Pulmonar Obstructiva Crónica (EPOC con FEV1 < 35% del
valor de referencia, PaO2 < 60 mm Hg o PaCO2 > 45 mm Hg).
- Riñón en herradura (no para AAT).
- Insuficiencia renal crónica en plan de diálisis.
- Insuficiencia hepática.
- Trasplante de órganos.
- Abdomen Hostil (pacientes con cirugías abdominales previas,
eventraciones, colostomías, ileostomías, etc. y/o abdomen irradiado).
- Tórax Hostil (con cirugía previas) o irradiado.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de procedimiento
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción de la endoprótesis
6. Comprobante de Implante
7. Protocolo del procedimiento
PROCESOS ARTERIALES: OBSTRUCTIVOS,
ESTENÓTICOS, ANEURISMÁTICOS, DEFORMATIVOS, CONGÉNITOS
Tecnología:
Tratamiento endovascular periférico por procedimientos
hemodinámicos, insumos (endoprótesis autoexpandibles, expandibles con
balón, o cubiertos/no cubiertos)
Fundamento terapéutico:
Se reconocerá el reintegro cuando se encuentre
imposibilitada la realización de cirugía a cielo abierto y cumpla con
alguno de los criterios predictores de riesgo:
• Infarto de miocardio agudo o reciente
con evidencia de riesgo isquémico determinadopor síntomas y/o estudios
no invasivos.
• Angina inestable (CF III o IV).
• Arritmias significativas: bloqueo AV de alto grado/arritmias
ventriculares sintomáticas/arritmias supraventriculares con ritmo
ventricular no controlado.
• Enfermedad valvular severa - Enfermedad Pulmonar Obstructiva Crónica
(EPOC conFEV1 < 35% del valor de referencia, Pa02 45 mm Hg).
• Riñón en herradura.
• Insuficiencia renal crónica en plan de diálisis.
• Insuficiencia hepática.
• Trasplante de órganos.
• Abdomen Hostil (pacientes con cirugías abdominales previas,
eventraciones,colostomías, ileostomías, etc. y/o abdomen irradiado).
• Acceso Hostil (con cirugía previas) o irradiado.
:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de procedimiento
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción del procedimiento
6. Comprobante de Implante
7. Protocolo del procedimiento
ESTENOSIS VALVULAR AÓRTICA
Tecnología: Válvula
aórtica protésica para implante percutáneo - TAVI
El reemplazo valvular aórtico percutáneo (RVAP) es un procedimiento que
consiste en lasustitución de la válvula aórtica nativa por una prótesis
acoplada o no, a un stent, que se colocapercutáneamente a través de vía
transarterial o transapical.
Fundamento diagnóstico:
Ecocardiograma transesofágico, Ecodoppler
color, Ergometría (si corresponde), Estudio de perfusión miocárdica (si
corresponde), Estudio Hemodinámico (si corresponde). Determinación del
riesgo quirúrgico por STS o Euroscore Logístico.
Fundamento terapéutico:
Pacientes con estenosis aórtica severa,
sintomática, que no puedan someterse a un reemplazo valvular
convencional, debido a un elevado riesgo quirúrgico (STS>10 % o
Euroscore logístico >20%), o pacientes en los cuales el reemplazo
valvular aórtico convencional se encuentre contraindicado por un equipo
quirúrgico. Los pacientes deben tener las siguientes condiciones:
• Anillo aórtico entre 18 y 29 mm
• Expectativa de vida mayor a 1 año (ausencia de enfermedades
terminales o severas sin posibilidad de curación)
• No presentar trombos en ventrículo izquierdo, endocarditis ni
condiciones con altoriesgo de obstrucción del ostium coronario
• No presentar válvula aórtica bicúspide
• Fracción de eyección mayor al 20%
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de procedimiento
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción del implante valvular
6. Ecocardiograma previo al
procedimiento
7. Comprobante de Implante
8. Protocolo del procedimiento
9. Información de resultados
o Mortalidad dentro de los 30 días
o Necesidad de marcapasos transitorio
o Otras complicaciones
o Clase funcional post-implante
INSUFICIENCIA CARDIACA AGUDA / SHOCK
CARDIOGENICO
Tecnología:
Dispositivo de asistencia ventricular (DAV) o corazón artificial y
módulo de seguimiento.
El Dispositivo de Asistencia Ventricular sustituye la función de bomba
de uno de los ventrículoso ambos. Estos dispositivos pueden ser
totalmente implantables o poseer además una unidad extracorpórea. El
fundamento para la utilización del DAV debe ser circunscripto
específicamenteen los casos en los cuales la función ventricular
deteriorada tiene probabilidad de recuperación o como puente al
trasplante cardíaco. Varios estudios han demostrado que las asistencias
ventriculares izquierdas se asocian a beneficios hemodinámicos,
neurohormonales y electrofisiológicos lo cual avala el uso del DAV como
puente a la recuperación ventricular.
Fundamento Terapéutico:
- Shock cardiogénico refractario al
soporte inotrópico a dosis máximas
y balón de contrapulsación (si no hay contraindicación) como puente
transitorio al trasplantecardíaco.
- Insuficiencia Cardíaca Aguda secundaria a Miocarditis aguda
fulminante como puente a la recuperación miocárdica.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de procedimiento
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción del dispositivo de
asistencia ventricular
6. Inscripción en lista de espera de
trasplante de INCUCAI
7. Comprobante de colocación del
dispositivo
8. Protocolo del procedimiento
9. Información de resultados
o Mortalidad dentro de los 30 días
o Otras complicaciones
10.
Resumen de Historia Clínica de seguimiento evolutivo en el caso del
módulo de seguimiento
INSUFICIENCIA CARDÍACA - INSUFICIENCIA
RESPIRATORIA
Tecnología:
Dispositivo de oxigenación por membrana extracorpórea (extracorporeal
membrane oxygenation - ECMO).
La ECMO constituye un sistema de soporte vital, el cual puede ser
utilizado en personas en estado crítico, con fallo pulmonar o
cardiopulmonar reversible, que no responden a técnicas o tratamientos
convencionales, lo cual puede incidir negativamente en el pronóstico
del paciente. Se define específicamente soporte cardiorrespiratorio
total o parcial durante un período suficiente, hasta que mejore la
patología que ocasiona el cuadro. Existen dos tipos de ECMO:
Veno-Venosa y Veno-Arterial.
Fundamento diagnóstico:
la valoración del equipo tratante constituye la
principal fuente en la que se basan y justifican la toma de decisiones.
Es importante tener en cuenta la recuperabilidaddel enfermo para no
realizar maniobras fútiles.
Fundamento terapéutico:
• Falla cardíaca: falla de salida de
circulación extracorpórea,
síndrome post-cardiotomía, falla aguda post IAM, miocarditis,
cardiomiopatías descompensadas, puente a asistencia ventricular,
trasplante o resucitación cardiopulmonar
• Falla respiratoria: Síndrome de distrés respiratorio del adulto,
síndrome de dificultad respiratoria del recién nacido, síndrome de
repercusión post trasplante pulmonar, crisis bronquial obstructiva
severa intratable y disnea secundaria a trauma.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de procedimiento
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción de la ECMO
6. Protocolo de ECMO
7. Inscripción en lista de espera de
trasplante de INCUCAI (en caso de ser usada como puente)
ARRITMIAS VENTRICULARES. PREVENCIÓN DE
MUERTE SÚBITA CARDÍACA
Tecnología:
Cardiodesfibrilador implantable uni o bicameral
El cardiodesfibrilador implantable (CDI) es un dispositivo con función
de estimulación Cardíaca similar a la del marcapasos. Lleva incorporada
la capacidad de detectar arritmias cardíacas que ponen en riesgo la
vida (taquicardia o fibrilación ventricular) y en esas circunstancias,
administrar un choque eléctrico, restaurando el ritmo normal del
corazón y evitando la muerte súbita por arritmia. El CDI consta de 1 o
2 catéteres que alcanzan las cavidades derechas del corazón y una
unidad funcional. Básicamente este dispositivo capta señales
intracardiacas y lasanaliza según los parámetros programados,
discriminando si se corresponden con una taquiarritmia ventricular.
Fundamento terapéutico:
Prevención Secundaria:
• Paciente reanimado de taquicardia
ventricular sostenida (TVS) /
fibrilación ventricular (FV) con inestabilidad hemodinámica o Paro
Cardiorrespiratorio, independientemente de la etiología, pero fuera del
contexto de una causa reversible.
• Enfermedad cardíaca estructural, independiente del grado de deterioro
de la función sistólica ventricular izquierda (FSVI) y TVS espontánea,
ya sea hemodinámicamente estable o inestable.
• Síncope de origen no determinado con TV o FV clínicamente relevante y
sostenida y hemodinámicamente significativa inducida en el estudio
electrofisiológico.
Prevención Primaria:
• Fracción de eyección ventricular izquierda (FEVI) = 35% debido a
infarto de miocardio previo, al menos 40 días antes o miocardiopatía
dilatada (MCPD) no isquémica, clase funcional New York Heart
Association (NYHA) II o III, bajo tratamiento médico óptimo (debe
incluir salvo contraindicaciones inhibidores de enzima convertidora de
angiotensina —IECA—, betabloqueantes, antialdosterónicos).
• Disfunción ventricular Izquierda
debido a IAM previo por lo menos 40
días antes, FEVI =30%, clase funcional NYHA I - Displasia del
ventrículo derecho arritmogénica/cardiomiopatía, con uno o más factores
de riesgo para la muerte súbita.
• Síndrome QT largo, experimentando síncope y/o TV mientras recibe
betabloqueantes.
• Pacientes no hospitalizados como puente a recibir un trasplante
cardíaco.
• Síndrome de Brugada con síncope, TV sostenida u otros factores de
riesgo asociados (muerte súbita familiar, inducción TV/FV en estudio
electrofisiológico).
• TV polimórfica catecolaminérgica con síncope y/o TV sostenida
documentada durante tratamiento con betabloqueantes.
• La enfermedad de Chagas recibirá CDI, siguiendo los mismos
lineamientos de la miocardiopatía no isquémica.
• TV sostenida y sintomática en un niño o adulto con cardiopatía
congénita. - Síncope recurrente de origen desconocido en un niño o
adulto con cardiopatía congénita en presencia de disfunción ventricular.
• Miocardiopatía hipertrófica definida y alguno de los siguientes
antecedentes: 1) Antecedente personal de muerte súbita o taquicardia
ventricular sostenida; 2) Historia de muerte súbita relacionada a la
miocardiopatía hipertrófica (MCH) en al menos un familiar en primer
grado; 3) Haber experimentado al menos un episodio sincopalreciente; 4)
Hipertrofia masiva del ventrículo izquierdo (espesor parietal máximo
igual o mayor de 30 mm); 5) Detección de taquicardia ventricular
sostenida (TVNS) en el registro ambulatorio de Holter; 6) Respuesta
hipotensiva o plana de la presión arterial durante el ejercicio
(incremento de la presión arterial menor a 25 mm Hg).
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de procedimiento
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción del CDI
6. Comprobante de colocación del CDI
7. Protocolo del procedimiento del CDI
ENFERMEDAD DE PARKINSON REFRACTARIA AL
TRATAMIENTO
Tecnología:
Neuroestimulador para neuroestimulación cerebral profunda (ECP).
La Neuroestimulación Cerebral Profunda es un tratamiento quirúrgico en
el cual se implanta un dispositivo, el neuroestimulador, que ha de
transmitir señales eléctricas a las áreas del cerebroque controlan el
movimiento, bloqueando las señales nerviosas anormales que generan el
temblor y otros síntomas invalidantes de la Enfermedad de Parkinson.
Este tipo de tratamientosólo se utiliza en los casos en que los
síntomas no puedan ser controlados con tratamiento medicamentoso
adecuado y en dosis óptimas. Este tratamiento no es curativo, pero
mejora el temblor, la rigidez, los movimientos lentos y las
dificultades motoras especialmente la marcha.
Fundamento Terapéutico:
Pacientes con Enfermedad de Parkinson avanzada
en quienes el tratamiento farmacológico a dosis óptimas resulte
insuficiente para el control de las variables motoras o cuando los
fenómenos de fluctuación resulten en un importante impacto en la
calidadde vida. No debe existir otro diagnóstico que pueda explicar la
no respuesta al tratamiento.
NO SE RECONOCERÁ EL REINTEGRO EN:
• Pacientes con severo déficit
cognitivo, demencia, atrofia cerebral o depresión que sería empeorado
por la ECP
• Psicosis, abuso de alcohol o abuso de drogas
• Edad mayor de 85 años
• Pacientes con Enfermedad de Parkinson Estadio V de Hoehn y Yahr que
es un estadio terminal caracterizado por caquexia, invalidez,
imposibilidad de pararse o caminar, querequiere constantes cuidados de
enfermería
• Lesiones estructurales como ACV de los ganglios basales, tumores, o
malformaciones vasculares como etiología de los trastornos del
movimiento
• Cirugía previa por trastornos del movimiento en los ganglios basales
afectados
• Discrasias sanguíneas
• Comorbilidades médicas, quirúrgicas, neurológicas que contraindiquen
la ECP.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de colocación del
Neuroestimulador
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción del Neuroestimulador
6. Comprobante de colocación del
Neuroestimulador
7. Protocolo del procedimiento de
colocación del Neuroestimulador
DOLOR CRÓNICO INTRATABLE
Tecnología:
Neuroestimulador espinal
La Neuroestimulación espinal comprende la estimulación de los cordones
posteriores de la médula. Consta de un generador de impulsos eléctricos
que se implanta en un bolsillo del tejido celular subcutáneo de la
región abdominal y, de electrodos que se disponen en el espacio
epidural unidos por un cable de conexión con el generador de impulsos
eléctricos.
Fundamento Terapéutico:
• Dolor Neuropático Crónico: se deberá
demostrar dolor durante un
tiempo establecido y el antecedente de ser refractario a por lo menos
cuatro (4) fármacos indicados para el dolor neuropático, de no existir
contraindicaciones, durante un tiempo no menor a tres 3 meses, no
habiendo disminuido el dolor más del 30% manteniéndose con una
intensidad no menor de 5 en la escala de 0 a 10 y generando una mala
calidad de vida.
• Dolor Lumbar: por lo menos seis (6) meses previos de dolor.
Procedimiento indicado en aquellos pacientes que han recibido varias
cirugías y en especial en aracnoiditis adhesivas o en el Síndrome de la
Cola de Caballo.
• Síndrome postIaminectomía.
• Síndrome de columna fallida.
• Síndrome doloroso regional complejo (SDRC) tipo I o tipo II.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de colocación del
Neuroestimulador
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción del Neuroestimulador
6. Comprobante de colocación del
Neuroestimulador
7. Protocolo del procedimiento de
colocación del Neuroestimulador
EPILEPSIA REFRACTARIA AL TRATAMIENTO
MÉDICO
Se define como la epilepsia resistente a fármacos en la que no se han
controlado las crisis tras el tratamiento adecuado con dos fármacos
antiepilépticos tolerados, adecuadamente elegidos y pautados
(monoterapia o en combinación), entendiendo como falta de control a la
aparición de crisis a lo largo de un año o que se presenten en un
tiempo inferior a tres veces el intervalo que mostraba antes de iniciar
el tratamiento.
Tecnología:
Neuroestimulador vagal
El Neuroestimulador vagal comprende la estimulación del nervio vago.
Consta de un generador de impulsos eléctricos que se implanta en un
bolsillo del tejido celular subcutáneo de la región infraclavicular y
de un electrodo helicoidal bipolar que se dispone alrededor del tramo
cervical del nervio vago izquierdo, y que está unido por un cable de
conexión con el generador.
Fundamento terapéutico:
• Diagnóstico de epilepsia confirmado
• Edad: 12 a 65 años
• Crisis parciales o generalizadas idiopáticas o de origen estructural
- Período interictal < 3 semanas - Fallo de la medicación tras 1 mes
de tratamiento comprobable con una o tres drogas con niveles estables y
máximos tolerados
• Cumplimiento adecuado del tratamiento farmacológico, documentando
debidamente su fracaso
• Convulsiones parciales que permanecen refractarias al tratamiento
óptimo con medicación antiepiléptica o tienen contraindicación o
intolerancia a todo tratamiento antiepiléptico, incluyendo el
tratamiento quirúrgico
No se reconocerá reintegro en:
• Vagotomía cervical previa
• Enfermedad neurológica progresiva o enfermedad sistémica
• Arritmias cardíacas
• Asma o EPOC
• Ulcera Péptica
• Diabetes insulino-dependiente
• Embarazo
• Historia de crisis no epiléptica.
Información requerida:
1. Fecha de Diagnóstico de la Patología
2. Fecha de colocación del
Neuroestimulador
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción del Neuroestimulador
6. Comprobante de colocación del
Neuroestimulador
7. Protocolo del procedimiento de
colocación del Neuroestimulador
Tecnología: Módulo de
cirugía para la epilepsia refractaria.
El módulo de cirugía para la epilepsia comprende los estudios invasivos
prequirúrgicos, el procedimiento quirúrgico propiamente dicho y el
material quirúrgico.
Se reconocen como procedimientos para la cirugía de la epilepsia
refractaria los siguientes: Hemisferectomía, Callostomía y Lobectomía
Temporal. Previo a la realización de la cirugía es necesaria la
evaluación del paciente: anamnesis y examen físico, detalle de las
características del síndrome convulsivo y su gravedad, circunstancias
sociales, examen neurológico, electroencefalograma de 24 horas para
registro de eventos interictales e ictales y estudios de neuroimagen
(RMN), etc.
Fundamento terapéutico:
diagnóstico definido de epilepsia y el cumplimiento de las siguientes
todas condiciones:
• Se ha descartado otra fuente de
ataques no epilépticos como los síncopes y las convulsiones psicógenas.
• Se documenta el diagnóstico de epilepsia claramente definido.
• En general, son candidatos los pacientes que presenten convulsiones
incapacitantes o toxicidad inaceptable por fármacos.
• La cirugía mejorará significativamente la calidad de vida.
• La frecuencia de las convulsiones interfiere la actividad diaria de
la persona.
• Debe haber pasado un periodo de tiempo adecuado con un uso correcto
de fármacos anticonvulsivantes en dosis adecuadas, con monitoreo de
estos en sangre y con la constancia de la adherencia al tratamiento.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de realización del
procedimiento
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción del procedimiento
6. Protocolo quirúrgico
AMPUTACIÓN DE MIEMBRO INFERIOR O
SUPERIOR
Tecnología: Prótesis para amputaciones de los miembros
Prótesis incluidas:
• Prótesis para desarticulación de
cadera con unidades de cadera y de
rodilla con sistemasde propulsión hidráulicos y/o neumáticos.
• Prótesis para amputación de miembro inferior sobre rodilla con
rodillas controladas por microprocesadores
• Prótesis para amputación supracondílea de miembro inferior con
unidades de rodillacon sistemas de propulsión hidráulicos y/o
neumáticos.
• Prótesis para amputación bajo rodilla con módulos y pie en fibra de
carbono, conos de siliconas, sistemas de vacío mediante bomba expulsora.
• Prótesis para amputación de miembro superior bajo codo con mano
mioeléctrica.
• Prótesis para amputación de miembro superior sobre codo con codo y
mano mioeléctrica.
Fundamento terapéutico:
Pacientes hasta 60 años con amputación de
miembro inferior, activosque ya hayan cumplido el tratamiento
preprotésico con buenos resultados demostrables de marcha sin requerir
asistencia mecánica o que hayan utilizado equipamiento previo con
resultados demostrables de marcha sin requerir asistencia mecánica.
Prótesis incluida:
- Prótesis para desarticulación de cadera con unidades de cadera y de
rodilla con sistemas de propulsión hidráulicos y/o neumáticos.
Fundamento terapéutico:
Pacientes hasta 50 años con amputación de
miembro inferior correspondiente según prótesis solicitada, activos que
ya hayan cumplido el tratamiento pre- protésico con buenos resultados
demostrables de marcha sin requerir asistencia mecánica o que hayan
utilizado equipamiento previo con resultados demostrables de marcha sin
requerir asistencia mecánica.
Prótesis incluidas:
- Prótesis para amputación de miembro
inferior sobre rodilla con rodillas controladas por microprocesadores.
- Prótesis para amputación supracondílea de miembro inferior con
unidades de rodillacon sistemas de propulsión hidráulicos y/o
neumáticos.
- Prótesis para amputación bajo rodilla con módulos y pie en fibra de
carbono, conos de siliconas, sistemas de vacío mediante bomba expulsora
Fundamento terapéutico:
Amputación o pérdida del miembro a nivel de la
muñeca o más arriba; imposibilidad de utilizar la prótesis estándar o
insuficiencia de ésta para satisfacer las necesidades funcionales del
paciente en la realización de actividades de la vida diaria;
preservación, en el muñón, de un umbral de microvoltios suficiente para
permitir el correcto funcionamiento de la prótesis; función
neurológica, miocutánea y cognitiva suficiente para manejar el
dispositivo; ausencia de comorbilidades que podrían interferir con el
mantenimientode la función de la prótesis; desempeño de actividades en
un entorno que no inhiba la función de la prótesis (por ejemplo, un
ambiente húmedo o descargas eléctricas puedan afectar el aparato);
superación de una prueba de control para ser considerado candidato.
Prótesis incluida:
- Prótesis para amputación de miembro
superior bajo codo con mano mioeléctrica.
Fundamento terapéutico:
Amputación o pérdida del miembro a nivel de la
muñeca o más arriba; imposibilidad de utilizar la prótesis estándar o
insuficiencia de ésta para satisfacer las necesidades funcionales del
paciente en la realización de actividades de la vida diaria.
Prótesis incluida:
- Prótesis para amputación de miembro
superior sobre codo con codo y mano mioeléctrica.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de provisión de la prótesis
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción de la Prótesis
6. Remito de provisión de la Prótesis
7. Conformidad del afiliado respecto
al elemento provisto
PRÓTESIS DE IMPLANTE TRAUMATOLÓGICAS
A. Prótesis de Cadera y/o Rodilla Oncológicas o no convencionales
Fundamento terapéutico:
Patología oncológica que lleve a la necesidad
de reemplazo de cadera. (Incluye la utilización de cemento quirúrgico
en cantidades y tipo que el cirujano utilice - asistencia técnica en el
quirófano - Set de colocación y extracción - Set completo en préstamo,
incluidas las sierras - Set de descartables: Steri-Drape, U-Drape -
hemosuctores y otros).
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de provisión de la prótesis
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción de la Prótesis
6. Protocolo quirúrgico
7. Comprobante de colocación de la
prótesis
B. Prótesis Especiales de Cadera - Primaria o de Revisión
(Incluyen: Modulares - Superficies cerámica- cerámica / cerámica -
polietileno o metal - metal/ de cabezas grandes 32mm / 36 mm/ 40 mm /
superficie metal trabeculadas).
Fundamento terapéutico:
Requerimientos específicos que determinen la necesidad de utilizar
prótesis especiales.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de provisión de la prótesis
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción de la Prótesis
6. Protocolo quirúrgico
7. Comprobante de colocación de la
prótesis
C. Prótesis Total Traumatológica
Oncológica o no convencional.
La prótesis incluye la articulación de la cadera, fémur completo y la
articulación de la rodilla.
Fundamento terapéutico:
Patología oncológica que lleve a la necesidad de reemplazo de cadera,
fémur y rodilla.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de provisión de la prótesis
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción de la Prótesis
6. Protocolo quirúrgico
7. Comprobante de colocación de la
prótesis
D. Instrumentación de Columna
Sistema de Instrumentación de Columna para Escoliosis: Evolutivas de
más de 30°(instrumentación por vía anterior o posterior) en pacientes
adultos o pediátricos.
Fundamento terapéutico:
Patología de escoliosis que requiera la tecnología específica.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de provisión de la prótesis
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción de la Prótesis
6. Protocolo quirúrgico
7. Comprobante de colocación de la
prótesis
HIPOACUSIA DE DIFERENTES ORÍGENES
Tecnología: Prótesis
implantable coclear y módulo de procedimiento quirúrgico de implante.
La prótesis para implante coclear es un dispositivo de alta tecnología
y precisión que está destinado a proporcionar o restablecer la audición
en aquellas personas que padecen hipoacusiade diferentes etiologías.
Consta de un dispositivo interno que se coloca bajo anestesia general
en el hueso temporal y ejerce la función de transductor del cual
emergen dos finos cables uno de masa, que queda anclado en el músculo
temporal y otro que lleva electrodos que se insertanen la rampa
timpánica de la cóclea. El dispositivo externo consta de un procesador
de sonidos y una bobina. El procesador capta los sonidos a través de un
micrófono, los transmite a un microprocesador que ejerce la función de
codificar la información sonora recibida y la transmitea la bobina.
Esta última se mantiene en relación con el dispositivo interno a través
de un campo magnético generado por un imán y transmite por
radiofrecuencia los sonidos codificados por elmicroprocesador
produciendo así la estimulación del nervio auditivo.
Fundamento Terapéutico:
PRELINGUALES:
• Desde los doce meses de edad con
hipoacusia perceptiva
(neurosensorial) profunda: pérdida de más de 90 db bilateral en las
frecuencias del habla (500 a 2000 cps).
• Desde los veinticuatro meses de edad con hipoacusia perceptiva
(neurosensorial) severa a profunda: pérdida de entre 60 y 90 db
bilateral en las frecuencias del habla (500 a 2000 cps).
• El uso de audífonos específicos (selección de audífonos) y
estimulación auditiva adecuada (respuesta a los audífonos) durante seis
meses, son requisitos previos necesarios, salvo casos de urgencia como
osificación coclear postmeningitis. Esta últimasustituye al primer
requisito.
• Prelinguales de más de seis años: los resultados dependen de las
adquisiciones lingüísticas previas al implante, es decir del grado de
oralización alcanzado. Evaluación según Categorías de Geers y Moog.
POSTLINGUALES:
• A cualquier edad hasta los sesenta
años según condición física
adecuada, sin contraindicaciones médicas ni psicológicas, con
hipoacusias severas a profundas con porcentajes de discriminación de
oraciones de hasta 50% con audífonos en el oído a implantar y con
audición residual de hasta 60% con audífonos en el oído contralateral.
• En hipoacusias progresivas el implante se indicará cuando las
evoluciones de las adquisiciones lingüísticas se vean limitadas de
acuerdo con la edad y la discriminación auditiva descienda de los
valores mencionados.
Las presencias de discapacidades concomitantes requieren evaluaciones
particulares para el Implante Coclear, algunas como la visual y sordera
puede ser prioridad uno, en otros casos con discapacidades motoras o
sensoriales asociadas el implante puede también ser prioritarios. A
pesar de mejorar la discapacidad global, en algunos casos el
aprovechamiento del implante es reducido.
Todo niño para implantar debe tener posibilidad de rehabilitación a
cargo de profesionales con capacitación adecuada, en su lugar de
residencia, rehabilitación que debe estar realizando, previamente al
implante, con audífonos.
Prestaciones incluidas:
Módulos para implantes de prótesis cocleares:
a)
Módulo
preimplante: comprende al menos los siguientes estudios:
• Audiometría
• B.E.R.A
• Entrenamiento en lectura labial en los casos que la requieran
• Estimulación eléctrica del promontorio - Evaluación psicológica -
Examen vestibular.
• Impedanciometría.
• Logoaudiometría.
• Otoemisiones acústicas.
• Selección de otoamplífonos.
• Timpanometría.
b)
Módulo Quirúrgico:
comprende al menos las siguientes prestaciones:
• Internación de adultos o pediátrica.
- Honorarios (de todo el equipo
profesional) y derechos operatorios, de anestesia, de monitoreo, de
oximetría de pulso, clínicos y de monitoreo del facial.
• Medicamentos y materiales descartables y quirúrgicos.
c)
Módulo de Seguimiento:
comprende las siguientes prestaciones, durante 3 (tres) meses
postquirúrgicos.
• Calibraciones (las necesarias),
incluyendo evaluación audiológica, de
percepción del habla y orientación familiar. Incluye supervisión del
profesional rehabilitador.
• Rehabilitación y adiestramiento auditivo.
• Eventualmente evaluación psicológica y apoyo psicoterapéutico.
1.
Fecha de Diagnóstico de la Patología
2. Fecha de provisión de la prótesis
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción del Implante Coclear
6. Protocolo quirúrgico
7. Comprobante de colocación del
Implante Coclear
8. Registro de calibraciones
Tecnología: Recambio
del procesador de la palabra
El procesador de la palabra es el componente externo necesario para el
funcionamiento del Implante Coclear. Consta de: micrófono (recoge los
sonidos) - microprocesador (procesa los sonidos codificándolos), bobina
(recibe los sonidos codificados por el microprocesador) y cable (por el
cual se conecta con la parte interna del implante) permitiendo que las
señales sonoras codificadas lleguen a los electrodos implantados y así
se pueda estimular el nervio auditivo.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de provisión de la prótesis
3. Matrícula de Profesional Tratante
4. Resumen de Historia Clínica que
justifique el uso de la tecnología
5. Prescripción del recambio del
procesador de la palabra
6. Remito de dispensa
FERTILIDAD
Tecnología:
Fecundación médicamente asistida de alta complejidad
Fundamento Terapéutico:
Persona mayor de edad.
Se encuentran incluidas en la presente Resolución las Técnicas de
Reproducción Médicamente Asistida de Alta Complejidad, las cuales
comprenden el proceso de Estimulación ovárica, Fecundación in vitro
(FIV), Inyección Intracitoplásmica de espermatozoides (ICSI) y
Transferencia de embriones.
El módulo comprende:
- Estudios de imágenes u otros estudios
de diagnóstico complementarios.
- Medicamentos utilizados para la inducción y/o preparación de la
gestación. Las drogas podrán incluir cualquiera de las siguientes:
Estrógenos; Derivados del Pregneno; Gonadotropina Coriónica Humana;
Hormona Foliculoestimulante (FSH); Coriogonadotropina alfa;
Corifolitrópina alfa; FSH- + Hormona Luteinizante; FSH + Lupotrina
Alfa; Clomifeno y los análogos de la GnRh (agonistas y antagonistas).
- Consultas profesionales de la totalidad del equipo interviniente y
procedimientos efectuados.
Sólo se dará curso a las solicitudes de reintegro presentadas por los
Agentes del Seguro que tengan por beneficiario a la mujer receptora de
los embriones, debiendo ser personas mayoresde edad en los términos que
determina la Ley N° 26.862.
Requisitos generales para acceder al
Reintegro:
Los Agentes del Seguro deberán presentar la siguiente información:
- Resumen de Historia Clínica en el
cual deberá constar de corresponder
antecedentespersonales, firmada por médico tratante y avalada por el
médico auditor con firma y sello.
- Listado completo de procedimientos, diagnósticos y terapéuticos
realizados, firmada pormédico tratante y avalada por el médico auditor
con firma y sello.
- Constancia de administración de las drogas con detalle del esquema
(dosis y número de aplicaciones) firmada por médico tratante y avalada
por el médico auditor con firma y sello.
- Documentación comprobatoria de embarazo y finalización de este.
- Constancia de inscripción de los prestadores en el Registro Federal
de Establecimientos deSalud
- REFES - del Ministerio de Salud de la Nación.
Información requerida:
1.
Fecha de transferencia embrionaria
2. Matrícula de Profesional Tratante
3. Prescripción del procedimiento de
reproducción medicamente asistida
4. Inscripción del centro en el REFES
5. Resumen de Historia Clínica que
justifique el uso de la tecnología
6. Protocolo de transferencia
embrionaria
7. Documentación Respaldatoria del
control de embarazo
8. Certificado de nacimiento
DROGODEPENDENCIA
Tecnología: Módulos
tratamientos de internación
a) Internación Psiquiátrica para
Desintoxicación: Intoxicación severa,
aguda, con descompensación clínico-psiquiátrica por uso indebido de
sustancias psicoactivas. Período máximo de otorgamiento: 30 días con
posibilidad de renovación por 30 días más, sujeto a la evaluación
profesional.
b) Internación en Comunidad
Terapéutica Residencial: En casos de
intoxicación crónica, severa con falta de contención familiar y cuando
no sostiene actividades laborales ni educativas. Deberán presentar
informes evolutivos mensuales por profesional tratanteperteneciente al
equipo profesional de la Institución prestadora y avalado por el médico
auditor. Período máximo de otorgamiento: 12 meses.
El
período máximo que se
reconocerá en concepto de reintegros para
tratamiento de Drogodependencia será
de
36 meses, sumadas todas las
modalidades requeridas para un mismo paciente. En caso de abandono de
tratamiento, se podrá renovar la prestación, por el término que reste
del módulo solicitado originalmente para ese paciente.
Se deberá adjuntar constancia de los días de internación cumplidos,
solo considerándose el módulo válido, cuando se constate más de
15 días
cumplidos por mes calendario.
Información requerida:
1.
Fecha de Diagnóstico de la Patología
2. Fecha de inicio de la internación
3. Matrícula de Profesional Tratante
4. Indicación médica de la internación
- Orden Judicial
5. Resumen de Historia Clínica que
justifique el uso de la tecnología
6. Constancia mensual de días de
internación cumplidos
7. Copia de inscripción de la
Institución en la Superintendencia de Servicios de salud.
DIABETES MELLITUS
Tecnología: Módulo de
seguimiento de pacientes con Diabetes Mellitus
Fundamento terapéutico:
Todo paciente con diagnostico confirmado de
Diabetes Mellitus en seguimiento por la Obra Social en un programa
integral de atención.
Información requerida:
Empadronamiento
del beneficiario:
1.
Resumen de Historia Clínica
2. Fecha de Diagnóstico de la Patología
3. Tipo de Diabetes (Tipo 1 - Tipo 2 -
Otros tipos)
4. Comorbilidades asociadas
o Dislipemia o Obesidad o Tabaquismo o
Hipertensión Arterial
5. Complicaciones de la DBT (puede ser
ninguna o todas)
o Hipertrofia del Ventrículo izquierdo
o Infarto de miocardio
o Insuficiencia cardiaca
o Accidente cerebrovascular
o Retinopatía
o Ceguera
o Neuropatía periférica
o Vasculopatía periférica
o Amputación de miembros
o Nefropatía
o Diálisis
o Trasplante Renal
6. Estudios complementarios
o Glucemia
o HbA1c
o LDL
o Triglicéridos
o Microalbuminuria
o TAS
o TAD
o Creatinina
o Peso
o Talla
o Circunferencia abdominal
o Fondo de ojos
o Inspección de pies
7. Tratamiento
o Antihipertensivos
o Hipolipemiantes
o Ácido Salicílico
o Hipoglucemiantes Orales
o Insulina
Seguimiento
del Beneficiario:
Actualización Anual
1.
Estudios complementarios
o Glucemia
o HbA1c
o LDL
o Triglicéridos
o Microalbuminuria
o TAS
o TAD
o Creatinina
o Peso
o Circunferencia abdominal
o Fondo de ojos
o Inspección de pies
2. Tratamiento
o Antihipertensivos
o Hipolipemiantes
o Ácido Salicílico
o Hipoglucemiantes Orales
o Insulina
HEMOFILIA A Y HEMOFILIA B
A.
Tratamiento a Demanda por
eventos y Profilaxis Intermitente
Fundamento terapéutico:
Pacientes con diagnóstico de Hemofilia A o B moderada o severa
Esta modalidad de tratamiento está destinada a pacientes con hemofilia
A, B y enfermedad de von Willebrand de cualquier gravedad que presenten
sangrado de cualquier localización (articular o extraarticular), se
encuentren o no bajo otra modalidad de tratamiento (profilaxis
primaria, secundaria o inmunotolerancia). A tales fines se aceptará la
provisión de stock para hacer frente a dicha demanda si los pacientes
sufrieran eventos frecuentes o la reposición si el evento requiera
tratamiento urgente, a fin de no demorar el tratamiento. La provisión
de stockdeberá responder al cálculo previo del requerimiento de
factores en los 6 meses anteriores y corresponderá a la cantidad máxima
promedio para dos meses de tratamiento, si bien podrán usarse en
tiempos mayores si no hubieran sido requeridos en ese tiempo.
La profilaxis intermitente está destinada a pacientes que:
- Fueran a someterse a intervenciones quirúrgicas menores o mayores o
procedimientos menores (extracciones dentarias, intervencionismo
diagnóstico, etc.) con la intención de prevenir sangrados.
- Hayan padecido una hemorragia grave y se utilizara para prevenir
nuevos sangrados
- Presenten hemartrosis a repetición cuando hubiera compromiso
articular demostrable por métodos imagenológicos
Esta modalidad será autorizada por un plazo de hasta 2 meses,
prorrogable otros dos meses contra actualización de resumen de Historia
Clínica y nuevas imágenes
Drogas incluidas:
- Factor VIII recombinante o de origen plasmático
- Factor IX recombinante o de origen plasmático
- Factor VIIa
- Complejo protrombínico activado (CCPA)
Empadronamiento
del beneficiario:
1.
Fecha de diagnóstico de la Patología
2. Matricula del Profesional Tratante
3. Fecha de inicio de tratamiento
sujeto a recupero
4. Documentación respaldatoria
o Resumen de historia clínica (Debe
estar detallado el Fundamento
Terapéutico y los antecedentes de medicación utilizada)
o Prescripción
de unidades utilizadas en el periodo
o Detalle de episodios de sangrado
o intervenciones
5. Información Adicional
o Peso
o Talla
o Valor de inhibidores
o Ha presentado sangrado en los últimos meses
- Articular (Número)
- Extraarticular (Número)
Seguimiento
del beneficiario:
Actualización Mensual o ante
presentación
1.
Matricula del Profesional Tratante
2. Documentación Respaldatoria
o Resumen de historia clínica (debe
comprender los nuevos episodios
presentados)
o Prescripción de unidades utilizadas en el periodo
o
Detalle de episodios de sangrado o intervenciones
3. Información Adicional
o Peso
o Talla
o Valor de inhibidores
o Presencia de sangrado en los últimos meses
- Articular (Número)
- Extraarticular (Número)
B.
Profilaxis primaria o
secundaria:
Fundamento Terapéutico:
Pacientes menores de 21 años en quienes se
hubiera sido establecido el diagnóstico de Hemofilia A o B severa
(dosaje de factor VIII o IX, respectivamentemenor al 1%), que hayan
tenido dos o más hemartrosis (secundaria o primaria respectivamente).
Para el caso de Hemofilia A, los pacientes que sean candidatos a esta
modalidad serán incluidosen la Compra Conjunta de Factor VIII-VIII
recombinante.
Drogas incluidas en el módulo
- Factor VIII recombinante o de origen plasmático
- Factor IX recombinante o de origen plasmático
- Factor VIIa*
- Complejo protrombínico activado (CCPA)*
- Emicizumab*
*Exclusivamente en pacientes con presencia de inhibidor circulante
mayor a 5 UB
Empadronamiento
del beneficiario:
1.
Fecha de diagnóstico de la Patología
2. Matricula del Profesional Tratante
3. Fecha de inicio de tratamiento
sujeto a recupero
4. Documentación respaldatoria
o Resumen de historia clínica (Debe
estar detallado el esquema de
profilaxis, el Fundamento Terapéutico y los antecedentes de medicación
utilizada)
o Prescripción de unidades utilizadas en el periodo
o
Detalle de episodios de sangrado o intervenciones
5. Información Adicional
o Peso
o Talla
o Valor de inhibidores
o Presencia de sangrado en los últimos meses
- Articular (Número)
- Extraarticular (Número)
Seguimiento
del beneficiario:
Actualización Mensual o ante
presentación
1.
Matricula del Profesional Tratante
2. Documentación respaldatoria
o Resumen de historia clínica (debe
comprender el esquema de profilaxis
y si ha presentado nuevos episodios)
o Prescripción de unidades
utilizadas en el periodo
o Detalle de episodios de sangrado o
intervenciones
3.
Información Adicional
o Peso
o Talla
o Valor de inhibidores
o Presencia de sangrado en los últimos meses
- Articular (Número)
- Extraarticular (Número)
C.
Inmunotolerancia
El tratamiento de inducción a la inmunotolerancia o inducción de
tolerancia inmunológica (ITI) es una modalidad de tratamiento basada en
la exposición al factor de la coagulación deficitario(usualmente el
VIII) de forma continua y más o menos intensiva, con el objetivo de
alcanzar la desensibilización del sistema inmune y permitir la
reutilización del factor deficitario como tratamiento sustitutivo.
Drogas incluidas en el módulo
- Factor VIII recombinante o de origen plasmático.
- Factor VIII con doble inactivación viral o recombinante.
Fundamento terapéutico:
pacientes con hemofilia A grave que han
desarrollado un inhibidormayor a 5 UB, demostrado en más de una
ocasión, y con demostración de interferencia en el resultado de la
profilaxis y el tratamiento de eventos hemorrágicos cuando son usadas
dosis estándar de factor VIII.
Dado que existen predictores de mala respuesta a la ITI, al inicio del
tratamiento se deberá consignar:
- Edad a la que se inicia el
tratamiento (edad superior a 8 años, se suele asociar a mala respuesta).
- Valor más alto de inhibidor previo al tratamiento (valor mayor a 10
UB, se asocia a mala respuesta).
- Pico histórico de inhibidor (mayor de 200 UB se asocia a mala
respuesta).
Los pacientes con dos factores predictores de mala respuesta podrán
iniciar con protocolos de dosis altas. En el caso de pico histórico
mayor de 200 UB, se aceptará protocolo de dosis altas.
El resto de los pacientes podrán iniciar con dosis bajas.
Las dosis intermedias se aceptarán en los casos de un predictor de mala
respuesta o una mala respuesta a dosis bajas con demostración efectiva
de frecuencia de sangrados u factores de mala respuesta intra
tratamiento.
Protocolos
- Dosis baja: 50 UI/kg cada 48 horas.
- Dosis intermedias: 100 UI/kg cada 24 horas.
- Dosis altas: 200 UI/kg cada 24 horas.
Tiempo máximo de tratamiento en caso de respuesta es de 33 meses.
En paciente de mal pronóstico, se podría considerar iniciar la ITI de
altas dosis con un factor VIII plasmático rico en factor von Willebrand
(VW) asociado.
Evaluación a los 6 meses Todos los pacientes deberán ser reevaluados
como mínimo a los 6 meses a fin de determinar factores pronósticos de
mala respuesta intratratamiento. La evaluación requerirá determinar:
- Si el título continúa aumentando
luego de 6 meses.
- Si el descenso del título es menor al 20%.
En ese caso se podrá pasar a protocolo de dosis intermedia si se
encontraba en dosis bajas,a dosis alta si estaba en intermedia o probar
cambio a factores con factor VW en caso de pacientes con dosis altas.
Se deberá evaluar la posibilidad de suspensión de la terapia en caso de
valores muy altos de inhibidor (mayor a 200 UB) luego de 6 meses.
Cada 6 meses será reevaluada la respuesta. En las evaluaciones
semestrales se considerará, además:
- Farmacocinética del Factor VIII luego
de su administración.
- Título de inhibidor.
- Cantidad de eventos en el período,
así como su gravedad y los factores/medicamentosy dosis requeridos para
tratarlos.
Empadronamiento
del beneficiario:
1.
Fecha de diagnóstico de la Patología
2. Matricula del Profesional Tratante
3. Fecha de inicio de tratamiento
sujeto a recupero
4. Documentación respaldatoria
o Resumen de historia clínica (Debe
estar detallado el esquema de
Inmunotolerancia, el Fundamento Terapéutico y los antecedentes de
medicación utilizada).
o Prescripción de unidades utilizadas en el
periodo.
o Detalle de episodios de sangrado
o intervenciones
o ausencia de episodios.
5. Información Adicional
o Peso
o Talla
o Valor de inhibidores
o Presencia de sangrado en los últimos meses
- Articular (Número)
- Extraarticular (Número)
Seguimiento del beneficiario:
Actualización Mensual o ante
presentación
1.
Matricula del Profesional Tratante
2. Documentación respaldatoria
o Resumen de historia clínica (debe
comprender el esquema de profilaxis
y si ha presentado nuevos episodios)
o Prescripción de unidades
utilizadas en el periodo
o Detalle de episodios de sangrado o
intervenciones
o Dosaje de inhibidor
3. Información Adicional
o Peso
o Talla
o Valor de inhibidores
o Presencia de sangrado en los últimos meses
- Articular (Número)
- Extraarticular (Número)
PACIENTES CON INTERNACIÓN EN
INSTITUCIONES DE REHABILITACIÓN DE ALTA COMPLEJIDAD (TERCER NIVEL)
Tecnología: Módulo de
internación en instituciones de rehabilitación de alta complejidad
(TercerNivel)
Fundamento terapéutico:
Pacientes cuya condición clínica justifique la
condición de internación en centros de rehabilitación de alta
complejidad en cualquiera de sus modalidades, incluyendo las
prestaciones de rehabilitación física, soporte nutricional, asistencia
de enfermería continua y asistencia respiratoria en caso de ser
requerido.
Los pacientes incluidos en el presente módulo no deberán poseer
Certificado Único de Discapacidad (CUD).
El recupero será de carácter mensual, y solo podrán ser incluidos a
recupero periodos completos del mes (30 días de prestaciones).
Se reconocerán hasta 6 meses del módulo por paciente.Módulos incluidos:
-
Módulo de paciente con requerimiento de ARM
- Módulo de paciente sin requerimiento
de ARM
Información requerida:
1.
Indicación del requerimiento de internación en Centro de rehabilitación.
2. Resumen de Historia Clínica donde
se consignen antecedentes
personales del paciente, tratamientos instituidos y resultados obtenidos
3. Detalle del módulo de internación
en Centro de rehabilitación, donde
conste: requerimiento y modalidad de rehabilitación, esquema de
seguimiento por parte de profesionales con detalle de prestaciones
realizadas contextualizadas en tiempo (diario, semanal, mensual),
requerimiento nutricional, medicamentos que se administran y
requerimiento de asistencia respiratoria.
MÓDULO DE PROFILAXIS PREEXPOSICIÓN
(PREP):
Se denomina profilaxis preexposición para el VIH (PrEP, del inglés Pre
Exposure Prophylaxis) al uso de medicación antirretroviral (ARV) en
personas no infectadas por el virus de inmunodeficiencia humana con el
objeto de reducir la posibilidad de infección. La PrEP implica el uso
(en general sostenido) de ARV antes y, de acuerdo con la estrategia,
después de la potencial exposición por vía sexual al virus.
PrEP es un componente de la Prevención combinada. Ésta última es una
estrategia que articula diferentes componentes de la prevención
(biomédico, comportamental y estructural), que se aplica en diferentes
niveles: de las personas, sus grupos sociales y vínculos, y su ámbito
social/territorial; con el objetivo de prevenir el contagio del HIV.
La Prevención combinada, que implica el uso de PrEP como una
herramienta clave, también incluye la provisión de preservativos y
geles lubricantes, testeo para VIH y otras ITS, y sus tratamientos,
educación sexual integral y acceso universal al tratamiento
antirretroviral en las personas que viven con HIV.
Se ha evaluado el PrEP a demanda, como una alternativa al PrEP diario,
empezando con 1 comprimido 24 horas antes y 48 horas después de la
situación de riesgo. Sin embargo, esta modalidad no ha sido adoptada en
todo el mundo como sí lo es el PrEP diario de manera continua.
Población objetivo:
- Varones cis que tienen relaciones
sexuales con otros varones cis y
mujeres trans que reporten uso inconsistente de preservativo en
relaciones sexuales anales (receptivas o insertivas) y/o hayan tenido
diagnóstico clínico de una ITS bacteriana en los últimos 6 meses y/o
hayan solicitado o recibido PrEP en más de una oportunidad.
- Parejas serodiscordantes en las que la persona HIV positiva no
mantenga una carga viral indetectable en forma sostenida y que reporten
uso inconsistente de preservativo en las relaciones sexuales.
- Trabajadores/as sexuales y/o personas en situación de prostitución
con uso inconsistente de preservativo.
- Usuarios/as de drogas que en los últimos seis meses hayan compartido
agujas u otros implementos.
- Personas que se auto perciben como de riesgo para infección por HIV.
Drogas incluidas en el módulo
- Tenofovir disoproxil
fumarato/Emtricitabina
- Tenofovir Alafenamida/Emtricitabina
- Tenofovir/Lamivudina
Información requerida:
Empadronamiento
del beneficiario:
1.
Resumen de Historia Clínica con indicación de PrEP
2. Matricula del Profesional Tratante
3. Fecha de inicio de tratamiento
sujeto a recupero
Actualización
Semestral
1.
Serología para HIV
2. Matrícula del Profesional Tratante
3. Actualización de la Información
Médica
o Cambio de tratamiento
o Fecha de finalización de tratamiento
o Motivo de Discontinuación
o En caso de progresión y/o cambio de tratamiento por favor adjuntar
nuevo resumen de Historia Clínica o RP del nuevo tratamiento
IF-2023-23380594-APN-SGE#SSS
ANEXO III
▪ Prácticas y Dispositivos médicos
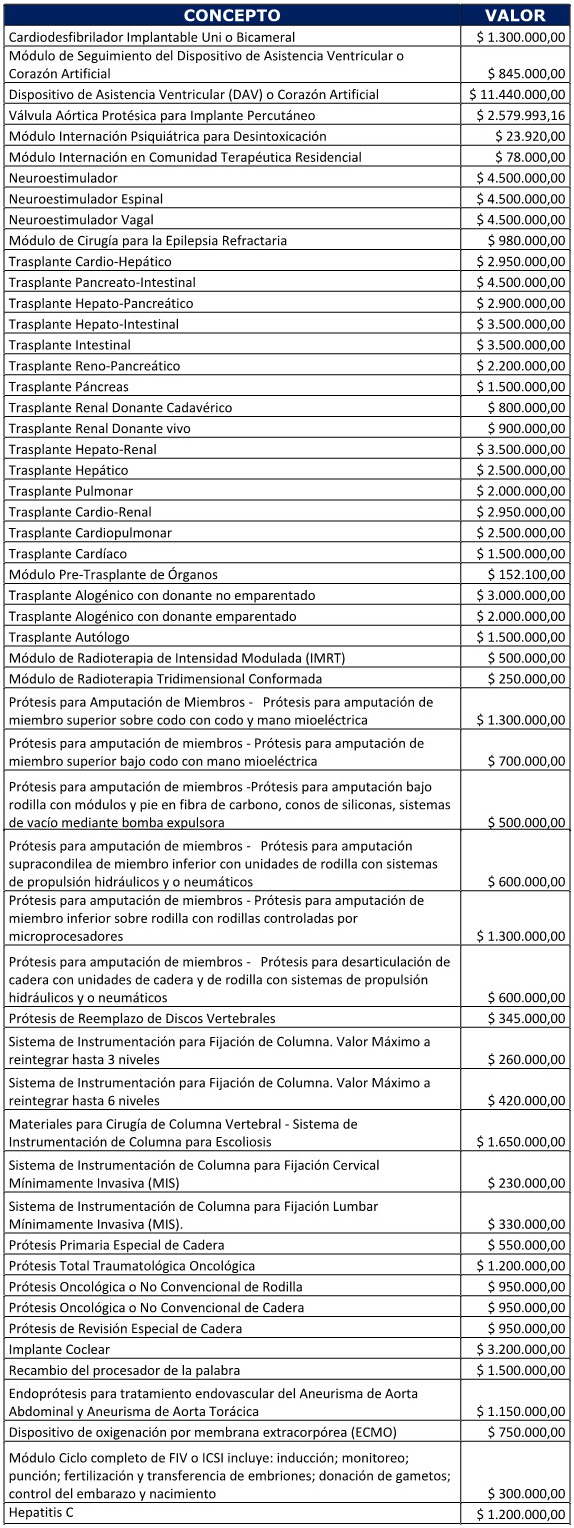
▪
Enfermedades Moduladas
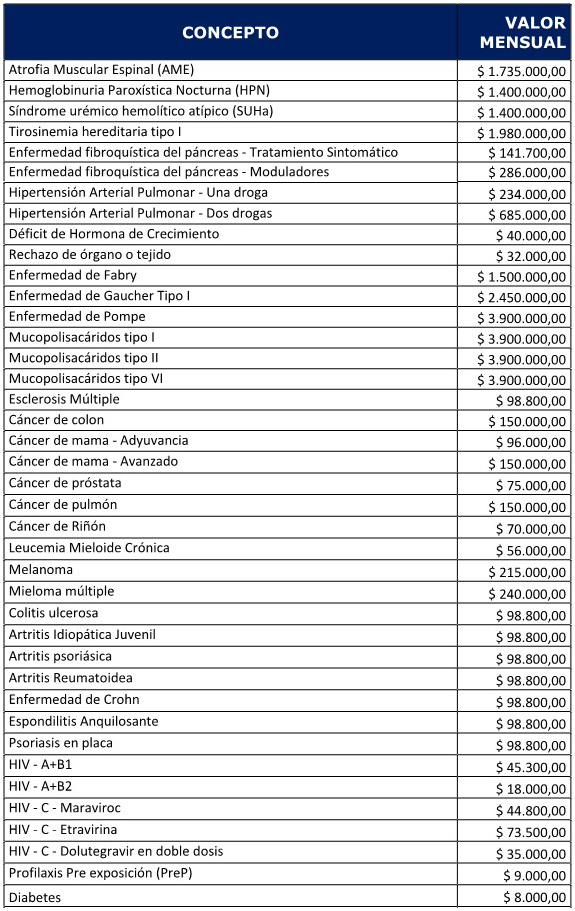
▪
Medicamentos para el tratamiento de Hemofilia Severa
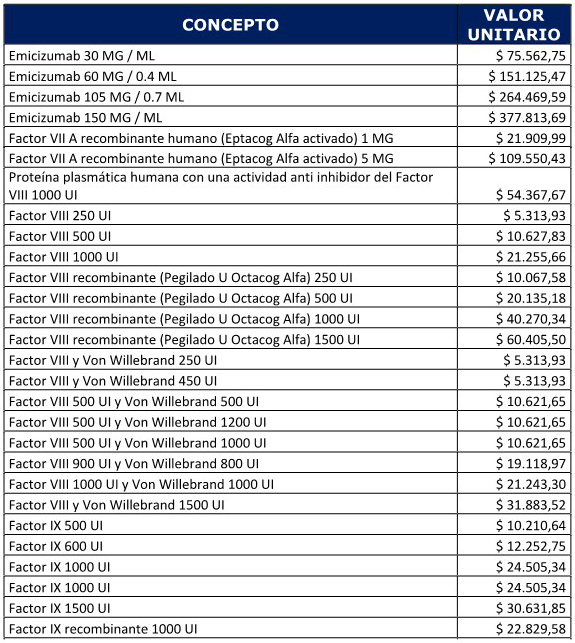
IF-2023-22932846-APN-SGE#SSS