
Servicio Nacional de Sanidad y Calidad Agroalimentaria
CONTROL DE PRODUCTOS DE ORIGEN ANIMAL
Resolución 138/2002
Requisitos que deberán cumplir los laboratorios que soliciten autorización para ingresar en la Red de Laboratorios del SENASA. Manuales de Calidad y de Procedimientos.
Bs. As., 25/1/2002
VISTO el expediente Nº 20.051/2001 del registro del SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA, las Resoluciones Nros. 1357 del 7 de diciembre de 1993 y 217 del 7 de abril de 1995, ambas del ex-SERVICIO NACIONAL DE SANIDAD ANIMAL, y
CONSIDERANDO:
Que es necesario actualizar los Requisitos de Antecedentes Analíticos que deben cumplir los Laboratorios que solicitan ingresar o mantenerse en la Red de Laboratorios SENASA, en rubros vinculados al control de productos de origen animal, clarificando la secuencia de estos procedimientos.
Que es necesario establecer los tiempos y condiciones de ingreso y conservación de las muestras a ser analizadas.
Que es necesario actualizar los Requisitos de Validación de Métodos Analíticos.
Que es propicio adecuar los procedimientos operativos vigentes como así también, verificar su implementación.
Que la Dirección de Asuntos Jurídicos ha tomado la intervención que le compete, no encontrando reparos de orden legal que formular.
Que el suscripto se encuentra facultado para el dictado del presente acto, conforme lo establecido por el artículo 8º, inciso n) del Decreto Nº 1585 de fecha 19 de diciembre de 1996, sustituido por su similar Nº 394 del 1º de abril de 2001.
Por ello,
EL PRESIDENTE DEL SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA
RESUELVE:
REQUISITOS ADMINISTRATIVOS.
Artículo 1º —(Artículo derogado por el art. 28 inciso a) de la Resolución N° 1446/2024 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 13/12/2024. Vigencia: a partir de su publicación en el Boletín Oficial)
Art. 2º — (Artículo derogado por el art. 28 inciso a) de la Resolución N° 1446/2024 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 13/12/2024. Vigencia: a partir de su publicación en el Boletín Oficial)
Art. 3º — (Artículo derogado por el art. 28 inciso a) de la Resolución N° 1446/2024 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 13/12/2024. Vigencia: a partir de su publicación en el Boletín Oficial)
Art. 4º — Los Manuales de Procedimientos deberán incorporar sin modificaciones (con excepción de Objeto, Alcance, Areas Afectadas, Responsabilidades y Relaciones que serán adaptados por cada Laboratorio de la Red, según su organigrama) los siguientes procedimientos establecidos por el SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA (SENASA), debiendo verificar el responsable de calidad del laboratorio el uso de la versión vigente.
17. Procedimiento para los Controles Intralaboratorios e Interlaboratorios vigencia a partir 1º de diciembre de 2001. (Anexo III, que forma parte de la presente resolución).
18. Procedimiento para Validación Interna de Métodos vigencia a partir del 31 de diciembre de 2001. (Anexo IV, que forma parte integrante de la presente resolución).
(Nota Infoleg: por art. 1° de la Disposición N° 125/2006 de la Dirección de Laboratorios y Control Técnico, B.O. 26/4/2006, se incorporaron nuevos "parámetros de validación". Vigencia: a partir de su publicación en el Boletín Oficial).
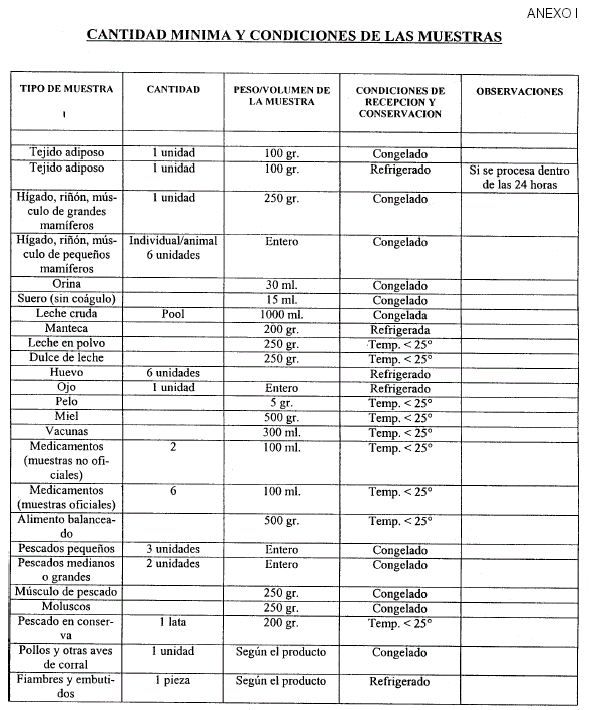
Art. 5º — La recepción y conservación de muestras debe cumplir los requisitos establecidos en el Anexo I, que forma parte integrante de la presente resolución.
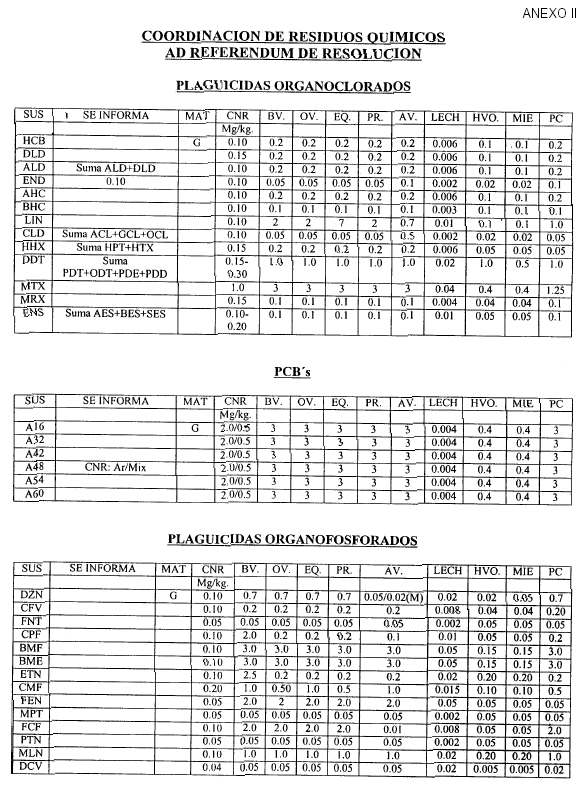
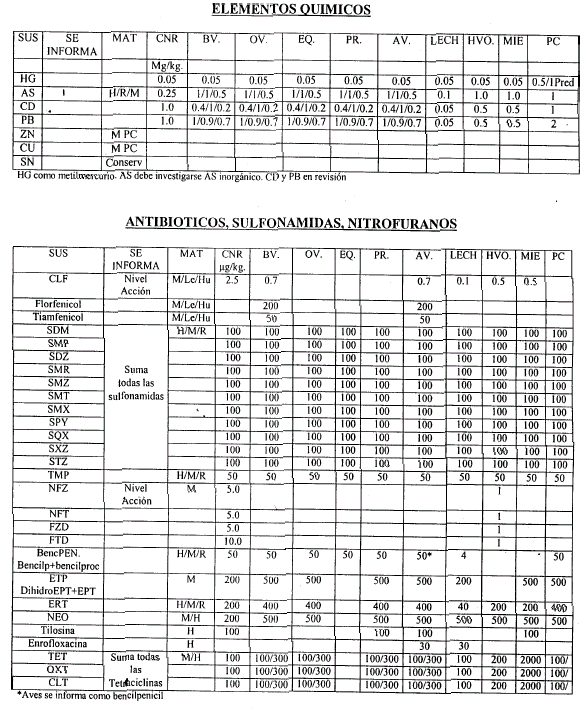
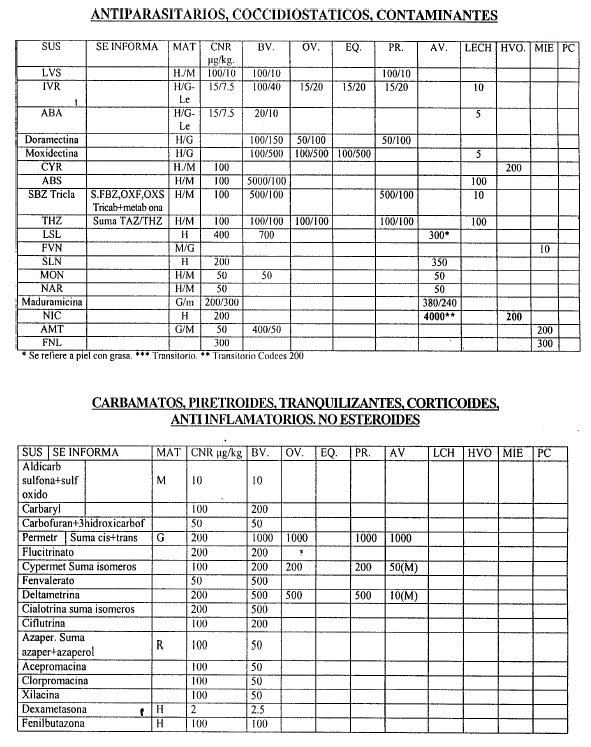
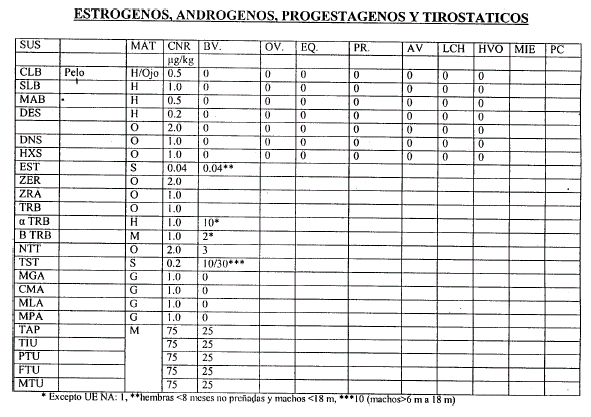
Art. 6º — Las validaciones Internas deben realizarse de acuerdo al Procedimiento indicado en el artículo 4º, considerando como orientación las Concentraciones Nominales de Referencia (CNR) que constan en el Anexo II, que forma parte integrante de la presente resolución, y que serán actualizados por la Coordinación de Residuos Químicos.
Art. 7º — Una vez analizadas las muestras deben conservarse de acuerdo a los siguientes requisitos:
a. Plaguicidas Organoclorados: se deben guardar las últimas SESENTA (60) muestras analizadas; los Positivos y Excesos se conservarán durante SEIS (6) meses. En todos los casos guardar una porción de grasa en rama y la alícuota fundida y filtrada.
b. Otros analitos: se deben guardar las últimas TRES (3) tandas o batches analizados. Los Excesos y Positivos se conservarán durante SEIS (6) meses.
Art. 8º — (Artículo derogado por el art. 28 inciso a) de la Resolución N° 1446/2024 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 13/12/2024. Vigencia: a partir de su publicación en el Boletín Oficial)
Art. 9º — (Artículo derogado por el art. 28 inciso a) de la Resolución N° 1446/2024 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 13/12/2024. Vigencia: a partir de su publicación en el Boletín Oficial)
Art. 10. — (Artículo derogado por el art. 28 inciso a) de la Resolución N° 1446/2024
del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O.
13/12/2024. Vigencia: a partir de su publicación en el Boletín Oficial)
Art. 11. —.- Derógase la Directiva de la Coordinación General del Laboratorio Animal de la Dirección de Laboratorios y Control Técnico (SENASA) Nº 1 del 17 de abril del 2001, a partir del 1º de diciembre de 2001 y las Directivas de la Coordinación de Residuos Químicos de la Coordinación General del Laboratorio Animal Nros. 1, 2 y 3 del 9 de abril de 1999, a partir del 31 de diciembre de 2001.
(Artículo sustituido por art. 1° de la Disposición N° 1/2002 Laboratorios y Control Técnico del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O.30/7/2002 Vigencia: a partir de su publicación en B.O.)
Art. 12. — La Dirección de Laboratorios y Control Técnico queda expresamente facultada para dictar las normas complementarias y/o modificatorias de la presente.
Art. 13. — La presente resolución entrará en vigencia a partir de su publicación en el Boletín Oficial.
Art. 14. — Comuníquese, publíquese, dése a la Dirección Nacional del Registro Oficial y archívese. — Bernardo G. Cané.
ANEXO III
CONTROLES INTRALABORATORIO E INTERLABORATORIO
1. OBJETO:
Establecer los requisitos que debe cumplir las muestras utilizadas para el control intralaboratorio e interlaboratorio de los métodos analíticos. Será adaptado por cada Laboratorio de la Red, según su organigrama.
2. ALCANCE:
Este procedimiento se aplica a todos los métodos analíticos implementados.
3. AREAS AFECTADAS:
Serán adaptadas por cada Laboratorio de la Red, según su organigrama.
4. RESPONSABILIDADES:
Profesionales y técnicos. Serán adaptadas por cada Laboratorio de la Red, según su organigrama.
5. RELACIONES:
Este procedimiento se vincula con Procedimientos de Calibración y Mantenimiento de Equipos, Procedimientos Analíticos. Serán adaptadas por cada Laboratorio de la Red, según su organigrama.
6. DESARROLLO:
6.1 Resumen:
Se establecen los requisitos de preparación y criterios que deben cumplir las muestras utilizadas para el control intralaboratorio en interlaboratorio de métodos analíticos.
6.2 Muestras Fortificadas:
6.2.1 Se define como muestra fortificada a una matriz de la misma naturaleza que las muestras a analizar enriquecida con una mezcla de uno o varios analitos cuya concentración sea mayor o igual que el mínimo nivel cuantificable y que se analiza con la tanda de muestras. Los resultados deben ser registrados y comparados con los valores nominales.
6.2.2 Debe registrarse la cantidad de matriz y solución patrón (identificando su lote y concentración) empleados para prepararla, el nombre del analista y la fecha de preparación. Esto rige tanto para las que se utilicen para la determinación de recuperaciones como para ensayos intralaboratorios y/o interlaboratorios.
6.3 Recuperaciones:
6.3.1 Se define como Recuperación Porcentual el cociente entre el valor obtenido de la concentración y la Concentración Nominal por CIEN (100), determinada a partir del análisis de una muestra de igual matriz que las muestras a analizar, enriquecida con una mezcla de los analitos en la Concentración Nominal de Referencia (CNR). Los porcentajes obtenidos para cada analito se usarán para corregir las concentraciones halladas de los mismos en las muestras de esa tanda. Para Residuos de Plaguicidas, PCB’s y Elementos químicos, cuando el método tenga una recuperación entre OCHENTA (80%) y CIENTO DIEZ (110%) POR CIENTO no debe ser corregido por recuperación.
6.3.2 Las Concentraciones Nominales de Referencia están relacionadas con el Limite Máximo de Residuos (LMR) o el Nivel de Acción (NA)y será tal que el MNC obtenido en la validación sea igual a la mitad o fracción menor del LMR.
6.3.3 Frecuencia de uso: mínimo UN (1) recuperado cada DIEZ (10) muestras o fracción menor. Excepto cuando se realiza de rutina gran cantidad de muestras, en ese caso UNO (1) cada VEINTE (20) muestras o fracción menor.
6.4 Límites de Recuperación:
6.4.1 Plaguicidas Organoclorados, PBC’s, Elementos Químicos: 70-110%. Ajustar la metodología para obtener un rango de 80-110%.
6.4.2 Plaguicidas Organofosforados, Antiparasitarios, Nitrofuranos, Sulfonamidas, Cloranfenicol: 60-110%.
6.4.3 Cuando la cuantificación se efectúa utilizando standard interno, la recuperación de los analitos deberá estar comprendida entre OCHENTA (80%) y CIENTO VEINTE (120%) POR CIENTO. Deberá determinarse la recuperación individual del standard interno y los límites de aceptación serán 60-110%. Cuando la recuperación del standard interno para una muestra no cumpla con el intervalo de aceptación deberá repetirse el análisis de la misma.
6.4.4 Para Inmunoanálisis:
6.4.4.1 Recuperación extractiva de 50-100% y recuperación global de 80-120%.
6.4.4.2 Para el caso de la determinación de hormonas en fluidos orgánicos, corregir los resultados si la recuperación de la corrida resulta menor que el 80%.
6.4.5 Cuando la recuperación de una tanda supere el Valor Promedio +/- 3 DS o se encuentre entre el Valor Promedio +/- 2 DS y +/- 3 D S por tercera vez consecutiva, se deberá repetir la corrida completa.
6.5 Gráficos de Control tipo X-R:
6.5.1 Construir los gráficos representando el valor promedio de las recuperaciones porcentuales diarias y estableciendo los Límites de Advertencia y de Control como Valor Promedio +/- 2 DS y Valor Promedio +/- 3 DS, respectivamente, determinados a partir de los valores históricos. Se considerarán como valores históricos los datos correspondientes a las últimas TREINTA (30) recuperaciones [IA últimas VEINTE (20) tandas], siempre que correspondan a un período no inferior a UN (1) mes.
6.5.2 Confeccionar las cartas de control para los Rangos de recuperaciones diarias, representando los Límites de Advertencia y de Control como Valor Promedio + 2 DS y Valor Promedio + 3 D S.
6.5.3 Si algún dato está fuera del límite de control hay que investigar la causa, tomar la acción correctiva correspondiente y documentarla. Los datos deben ser registrados diariamente.
6.5.4 Métodos multirresiduos: efectuar las cartas de control de aquellos analitos (3 ó 4) que sean más críticos (quedan retenidos en columna de purificación, se pierden por evaporación, son de incidencia más frecuente).
6.5.5 Inmunoanálisis: además deben confeccionarse las Cartas de Control para DB 50%,% Inespecífico, Pendiente y Recuperación global.
6.6 Muestras Intralaboralorio:
6.6.1 Muestras fortificadas que no son conocidos por el analista (muestras ciegas) y se utilizan como controles para que el supervisor verifique la calidad de los análisis que se están produciendo.
6.6.2 Frecuencia: Mínimo 1 mensual por analista/matriz/analito.
6.6.3 Planilla de registro. Se debe dejar constancia de fecha de análisis, nombre del analista, identificación del analito, concentración nominal, concentración hallada, porcentaje de recuperación de la tanda, concentración hallada corregida por recuperación, diferencia porcentual con respecto a la concentración nominal, firma del supervisor, observaciones.
6.6.4 Criterio de aceptación de acuerdo a la concentración de analito.
|
CNR |
Cv% |
DIFERENCIA RESPECTO VN |
|
>100ppb |
<12% |
+/-15% |
|
10 a 100 ppb |
<17% |
+/-20% |
|
< 10 ppb |
<21% |
+/-25% |
|
< 1 ppb |
<30% |
-30% +20% |
|
IA 1 a 3 ppb |
<14% |
-25%+20% |
|
IA 0.5 a 1 y 3 a 5 ppb |
<20% |
-30%+20% |
(Cuadro sustituido por art. 2° de la Disposición N° 1/2002 Laboratorios y Control Técnico del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O.30/7/2002. Vigencia: a partir de su publicación en B.O.)
6.6.5 En "Observaciones" señalar las acciones correctivas implementadas cuando no se cumple el criterio de aceptación.
6.6.6 Cuando se hayan efectuado DIEZ (10) determinaciones se deben promediar los valores obtenidos y hallar CV% que debe cumplir con el criterio antes definido.
6.7 Muestras Interlaboratorio:
Se realizarán en acuerdo con las Normas ISO / IEC 43-1 O IRAM 305-1 e IRAM 305-2:
• Proceso de evaluación, Prueba de aptitud, Programa de ensayos interlaboratorios (Anexo II).
Proceso de evaluación, Prueba de aptitud, Programa con valores conocidos (Anexo III)
6.7.1 Rondas organizadas y evaluadas por DILACOT. Deben conservar ordenados por fecha:
6.7.1.1 Resultados de los análisis de las muestras correctamente identificadas.
6.7.1.2 Información remitida a DILACOT con copia de los registros.
6.7.1.3 Informe final enviado por DILACOT.
6.7.2 Criterio de aceptación: no falsos positivos ni negativos, diferencias:
|
Concentración de analito |
Concentración corregida por recuperación |
Concentración sin corregir por recuperación |
Confirmación por HPLC-IA |
|
> 100 ppb |
+/-20% |
Entre - 20% y+ 10% |
+/-25% |
|
> 10 y <100 ppb |
+/-25% |
Entre - 25% y + 20% |
+/-30% |
|
<10 ppb |
+/-30% |
Entre - 30% y +20% |
+/-35% |
|
<1 ppb |
- 30% y +20% |
|
|
|
> 1 y < 3 ppb (por IA) |
- 30%/+20% |
Entre - 35% y +20% |
-35%/+25% |
|
>0.5 y< 1 ppb, >3 y < 5 (por IA) |
- 35%/+20% |
Entre - 40% y +20% |
- 40%/ + 25% |
(Cuadro sustituido por art. 3° de la Disposición N° 1/2002 Laboratorios y Control Técnico del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O.30/7/2002. Vigencia: a partir de su publicación en B.O.)
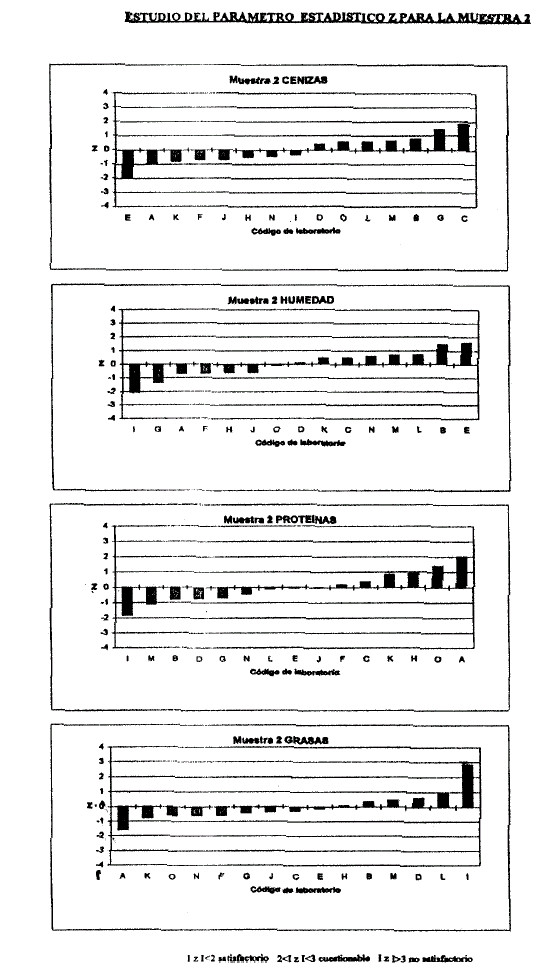
6.7.3 Si la cantidad de Laboratorios participantes lo permite se realizará el análisis estadístico Puntaje Z (Anexo IV, V).
Z= (x-X)/DS
• x: valor obtenido.
• X: valor asignado o verdadero.
• DS: desvío standard.
Si: Z< 2: Satisfactorio. 2<Z <3: Condicional. Z> 3: No satisfactorio.
Para mantenerse en la Red de Laboratorios debe obtener como mínimo OCHENTA (80%) POR CIENTO Satisfactorio, VEINTE (20%) POR CIENTO Condicional y ningún No Satisfactorio.
6.7.4 Frecuencia mínima 2 anuales de los cuales uno podrá ser organizado por un organismo internacional reconocido previamente por DILACOT-SENASA.
6.8 Muestras de control tomadas en inspección:
Muestras de control: Muestra de control remitidas a la Dirección de Laboratorio y Control, Técnico o contramuentra que queda en Inspección Veterinaria, y/o aquellas tomadas en los laboratorios durante una inspección de rutina, al ser analizadas en DILACOTE no deben presentar:
a- falsos positivos y falsos negativos.
b- Resultados cuantitativos que difieren: en más de TREINTA POR CIENTO (30%) para valores de concentración mayores que 100 ppb, en más de TREINTA Y CINCO POR CIENTO (35%) para concentraciones entre 10 y 100 ppb y en más de CUARENTA POR CIENTO (40%) para concentraciones menores que 10 ppb.
7. REGISTRO Y ARCHIVO:
Conservar como mínimo durante SEIS (6) años. Interlaboratorios es conveniente conservarlos DIEZ (10) años.
(Item 7 sustituido por art. 1° de la Disposición N° 6/2004 de la Dirección de Laboratorios y Control Técnico B.O. 30/11/2004. Vigencia: a partir de su publicación en el Boletín Oficial).
8. ANEXO:
ANEXO I: Lista de Distribución.
ANEXO II: Proceso de Evaluación, Prueba de Aptitud, programa de Ensayos InterIaboratorios.
ANEXO III: Proceso de Evaluación, Prueba de Aptitud, Programa con Valores Conocidos.
ANEXO: Normas ISO/IEC 43-1 o IRAM 305-2
9. HISTORICO:
No contiene.
10. REFERENCIAS:
AOACI Quality Assurance Principies. F. Garfield 1994, The International Harmonized portocol for Proficiency Testing of Analytical Laboratories, IUPAC 1993, y Directivas de la Coordinación de Residuos Químicos.
ANEXO I
LISTA DE DISTRIBUCION
|
NOMBRE |
TIPO DE COPIA |
FIRMA |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
........................................................
Firma Responsable - Aclaración


(Anexo sustituido por art. 4° de la Disposición N° 1/2002 Laboratorios y Control Técnico del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O.30/7/2002. Vigencia: a partir de su publicación en B.O.)

ANEXO III

(Anexo sustituido por art. 5° de la Disposición N° 1/2002 Laboratorios y Control Técnico del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O.30/7/2002. Vigencia: a partir de su publicación en B.O.)
ANEXO IV
REQUERIMIENTOS PARA LA VALIDACION INTERNA DE METODOS ANALITICOS Y PRESENTACION DE INFORMES DE VALIDACION
1. OBJETO:
Estandarizar los requisitos, parámetros, criterios, diseño y presentación de informes de los procedimientos de validación interna de metodologías analíticas.
2. ALCANCE:
Validación interna de métodos analíticos utilizados para el control de alimentos y residuos químicos.
3. AREAS AFECTADAS:
Serán adaptadas por cada Laboratorio de la Red, según su organigrama.
4. RESPONSABILIDAD:
Es responsabilidad de los Jefes de Departamento, Responsables de Programas o Director Técnico.
Serán adaptadas por cada Laboratorio de la Red, según su organigrama.
5. RELACIONES:
No indicadas a la fecha.
6. DESARROLLO:
6.1 Definiciones y/o abreviaturas:
6.1.1 ANALISIS DE PERICIA (PROFICIENCIA): Una evaluación periódica de la performance de laboratorios individuales y grupos de laboratorios. Se lleva a cabo cuando un Organismo independiente distribuye materiales típicos para análisis, sin supervisión por parte de los participantes.
[IUPAC Orange Book]
6.1.2 ANALIZADO: Cantidad particular sujeta a medida.
Nota: la especificación de un analizado puede requerir declaraciones acerca de las magnitudes, tales como tiempo, temperatura y presión.
[VIM 1993]
6.1.3 ASEGURAMIENTO DE CALIDAD: Son todas las actividades planificadas y sistemáticas puestas en marcha dentro del sistema de calidad, y demostradas como necesarias, para brindar la confianza adecuada de que una entidad cumplirá con los requisitos de calidad.
[ISO 8402: 1994]
6.1.4 CALIDAD: Son todas las cualidades y características de un producto o servicio que tienen capacidad para satisfacer los requisitos establecidos.
[ISO 8402: 1994]
6.1.5 CONTROL DE CALIDAD: Técnicas y actividades operativas que se usan para cumplir con los requisitos de calidad.
[ISO 8402: 1994]
6.1.6 CONTROL INTERINO DE CALIDAD: Es una serie de procedimientos que lleva a cabo el personal de un laboratorio para el monitoreo continuo de las operaciones y los resultados de las medidas, a fin de decidir si los resultados son lo suficientemente confiables para ser liberados.
[IUPAC Orange Book]
6.1.7 CURBA DE CALIBRACION: Es la representación gráfica de la señal de medida como una función de cantidad de analito.
[AOAC-PVMC]
6.1.8 DESVIACION: Diferencia entre los resultados de los análisis y el valor de referencia aceptado.
Nota: la desviación es un error sistemático total en contraposición al error aleatorio. Puede haber UNO (1) o más componentes de error sistemático que contribuyen a la desviación. Una diferencia sistemática importante en relación al valor de referencia aceptado, se refleja en un mayor valor de sesgo.
[ISO 3534-1]

6.1.11 DESVIACION ESTANDAR DE LA REPRODUCIBILIDAD: La desviación estádar de los resultados de análisis obtenidos bajo condiciones de reproducibilidad.
Nota: esta es una medida de dispersión de la distribución de resultados de análisis bajo condiciones de reproducibilidad. Similarmente la variación de reproducibilidad y coeficiente de reproducibilidad, se podría definir y usar como medidas de dispersión de resultados de análisis bajo condiciones de reproducibilidad.
[ISO 3534-1]
6.1.12 DISCRIMINACION: Es la capacidad de un instrumento de medición para responder a pequeños cambios en el valor del estímulo.
[VIM 1984]
6.1.13 ERROR (DE MEDIDA): Es el resultado de una medida menos el valor verdadero del analizado.
Nota: dado que no se puede determinar un valor verdadero, en la práctica se usa un valor verdadero convencional.
[VIM 1993]
El valor de un resultado menos el valor verdadero.
[IUPAC Compendium of Chemical Technology, 1985]
6.1.14 ERROR ALEATORIO: Resultado de una medida menos la media que podría resultar de un número infinito de medidas del mismo analizado, llevado a cabo bajo condiciones de repetibilidad.
Nota: el error aleatorio es igual al error menos el error sistemático. Dado que se puede hacer sólo un número finito de medidas, es posible determinar solo una estimación de error aleatorio.
[VIM 1993]
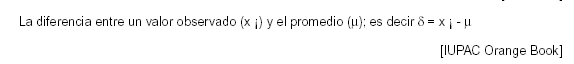
6.1.15 ERROR SISTEMATICO: E s la media que resultaría de un número infinito de medidas del mismo analizado, llevado a cabo bajo condiciones de repetibilidad menos el valor verdadero del analizado.
Nota: el error sistemático es igual al error menos el error aleatorio. Como el valor verdadero, el error sistemático y sus causas no se pueden conocer.
[VIM 1993]
6.1.16 ESPECIFICIDAD: La capacidad del método para medir sólo lo que se propone medir.
[AOAC-PVMC]
La especificidad es la capacidad para evaluar sin equivocación el analito en la presencia de componentes que se espera que puedan estar presentes. Típicamente estos podrían incluir impurezas, productos de degradación, matrices, etc.
[ICH Q2A, CPMP/ ICH/381/95]
6.1.17 EVALUACION DE LA ROBUSTEZ (RUGGEDNESS): Estudio intralaboratorio para comprobar el funcionamiento de un proceso analítico cuando se realizan pequeños cambios en el medio ambiente y/o condiciones operativas, semejantes a aquellos que probablemente surjan en diferentes medio ambientes de análisis. La evaluación de robustez permite obtener información de los efectos producidos por cambios menores, en forma rápida y sistemática.
[AOAC-PVMC]
6.1.18 EXACTITUD: Es la cantidad que se refiere a las diferencias entre la media de una serie de resultados o un resultado individual y el valor que se acepta como verdadero o valor correcto, para la cantidad medida.
[IUPAC Compendium of Chemical Technology, 1985]
6.1.19 EXACTITUD (DEL INSTRUMENTO DE MEDIDA): Es la capacidad de un instrumento de medida para dar respuestas a un valor verdadero.
Nota: en este contexto, la exactitud es un concepto cualitativo.
[IUPAC Orange Book]
6.1.20 FALSOS NEGATIVOS/POSITIVOS: Para los métodos cualitativos, se puede determinar la proporción de falsos positivos/negativos. Se deben brindar datos de una comparación de método confirmatorio, si tal o tales métodos son aplicables a las mismas matrices y al mismo rango de concentración. En la ausencia de un método para realizar la comparación, se deben analizar las poblaciones de muestras negativas o positivas fortificadas. Se pueden determinar los falsos positivos/negativos como sigue a continuación:
Proporción falso positivo (%)= falso positivo x 100/total negativos conocidos
Proporción falso negativo (%)= falso negativo x 100/total positivos conocidos
[AOAC Research-Institute - Performance tested Methods Programme, Procedure]
Falso Positivo: Probabilidad que la concentración real de un analito en la muestra de laboratorio sea menor que el valor límite aceptado (LMR o NA) mientras que las medidas realizadas en UNA (1) o más porciones de muestra, indican que la concentración excede ese valor. Los valores aceptados para esta probabilidad están en el rango de UNO (1) a CINCO POR CIENTO (5%).
[Codex Alimentarius, ALINORM 01/24A]
Falso Negativo: Probabilidad que la concentración real de un analito en la muestra de laboratorio sea mayor que el valor límite aceptado (LMR o NA) mientras que las medidas realizadas en UNA (1) o más porciones de muestra, indican que la concentración no excede ese valor. Los valores aceptados para esta probabilidad están en el rango de UNO (1) a CINCO POR CIENTO (5%).
[Codex Alimentarius, ALINORM 01/24A]
6.1.21 INCERTIDUMBRE (DE MEDIDA): Es el parámetro asociado con el resultado de una medida, que caracteriza la dispersión de los valores que se pueden atribuir razonablemente al analizado.
Nota: El parámetro puede ser, por ejemplo, una desviación estándar (o múltiplo dado de ella) o la amplitud de un intervalo de confianza. La incertidumbre de medidas comprende, en general, muchos componentes. Algunos de estos componentes se pueden evaluar a partir de la distribución estadística de los resultados de una serie de medidas y se pueden caracterizar por desviaciones estándar experimentales. Los otros componentes que también se pueden caracterizar por desviaciones estándar, se evalúan de distribuciones de probabilidad asumidas, basadas en la experiencia u otra información. Se entiende que el resultado de la medida es la mejor estimación del valor del analizado y que todos los componentes de incertidumbre, que incluyen aquellos que surgen de efectos sistemáticos, tales como componentes asociados con las correcciones y estándares de referencia, contribuyen a la dispersión.
[VIM 1993]
6.1.22 LIMITE DE CUANTIFICACION: Es el contenido igual o mayor que el menor punto de concentración de la curva de calibración.
[AOAC-PVMC]
También llamado Límite de informe.
Es la concentración mínima de un analito que puede determinarse con precisión aceptable (repetibilidad) y exactitud, bajo las condiciones establecidas en el análisis.
[NATA-Note #13]
También llamado Límite de cuantificación.
Los límites de cuantificación son las características de realización que marcan la capacidad de un proceso de medidas químico para cuantificar el analito adecuadamente.
Nota: La capacidad para cuantificar se expresa generalmente, en términos de señal o valor de analito (verdadero) que producirá estimación, teniendo desviación estándar relativa especificada (RSD), del DIEZ POR CIENTO (10%) o VEINTE POR CIENTO (20%), según el nivel de concentración del analito.
Por consiguiente: LQ = kQ sQ, donde LQ es el límite de cuantificación, sQ es la desviación estándar en ese punto y kQ es el factor cuya inversa multiplicada por el RSD iguala al cuantificando seleccionado. El valor por defecto que expresa IUPAC para kQ es 10.
[IUPAC Orange book]
(Nota sustituida por art. 2° de la Disposición N° 6/2004 de la Dirección de Laboratorios y Control Técnico B.O. 30/11/2004. Vigencia: a partir de su publicación en el Boletín Oficial).

6.1.24 LINEALIDAD: Define la capacidad del método para obtener los resultados de los análisis proporcionales a la concentración de analito.
Nota: El Rango Lineal es por inferencia del rango de concentraciones de analito en el cual el método da resultados del análisis proporcional a la concentración del analito.
[AOAC-PVMC]
6.1.25 MATERIAL DE REFERENCIA (RM): Es el material o sustancia en el cual UNO (1) o más valores de sus propiedades son suficientemente homogéneos y están bien definidos para permitir utilizarlos para la calibración de un instrumento, la evaluación de un método de medición o la asignación de valores a los materiales.
Nota: Un material de referencia describe materiales que a menudo se llaman estándares de medida, por ejemplo, sustancias químicas usadas para calibración o identificación. Es necesario tener cuidado cuando se usa el término estándar dado que comúnmente se lo utiliza en DOS (2) contextos diferentes. El término puede referirse a los estándares de la medida en el sentido de material de referencia, o puede referirse a normas escritas, tales como métodos estándar. Es importante asegurarse de que esta diferencia sea clara.
[ISO/IEC Guide 30-1992, 2.1]
6.1.26 MATERIAL DE REFERENCIA CERTIFICADO (CRM): Es el material de referencia acompañado de un certificado, en el cual UNO (1) o más valores de sus propiedades están certificados por un procedimiento que establece su trazabilidad con una realización exacta de la unidad en la que se expresan los valores de la propiedad, y para la cual, cada valor certificado se acompaña de una incertidumbre, con la indicación de un nivel de confianza.
[ISO/IEC Guide 30-1992, 2.2]
6.1.27 MEDIDA: Sede de operaciones que tienen por objeto determinar el valor, de una cantidad.
[VIM 1993]
6.1.28 METODO DE MEDIDA: Es la secuencia lógica de operaciones, descriptas genéricamente, usadas en la realización de medidas.
[VIM 1993]
6.1.29 PRECISION: Es la proximidad entre resultados de análisis independientes obtenidas bajo condiciones estipuladas.
Nota: La precisión depende sólo de la distribución de los errores aleatorios y no se relaciona con el valor verdadero o valor especificado. La medida de precisión se expresa usualmente en términos de imprecisión y está computada como desviación estándar de los resultados de los análisis. Los resultados de los análisis independientes son los resultados obtenidos de forma tal que no esté influenciado por ningún otro resultado previo del mismo o similar objeto de análisis. Las medidas cuantitativas de precisión dependen críticamente de las condiciones estipuladas. La repetibilidad y reproducibilidad son series particulares de condiciones extremas.
[ISO 3534-1]
6.1.30 PROCEDIMIENTO DE MEDIDA: Serie de operaciones, descriptas específicamente, usadas para realizar medidas de acuerdo con un método dado.
Nota: un procedimiento de medida normalmente se registra en un documento que a veces en sí mismo, un procedimiento de medida o método de medida y tiene usualmente suficiente detalle para permitir al operador, llevar a cabo una medida sin información adicional.
[VIM 1993]
6.1.31 RANGO (MEDICION Y TRABAJO): Es una serie de valores analizados para los que se pretende que el error de un instrumento de medida, esté dentro de los límites especificados.
[IUPAC Orange Book]
6.1.32 REACTIVIDAD CRUZADA: Respuesta (del método) a análogos, metabolitos u otros componentes no objetivos que puedan estar presentes en las matrices.
[AOAC-PVMC]
6.1.33 RECUPERACION: Es la fracción del analito agregada a la muestra (muestra fortificada) antes del análisis, analizadas muestras fortificadas y sin fortificar, el porcentaje de recuperación (%R) se calcula como sigue:
%R = [(CF-CU)/CA] x 100
Donde CF es la concentración de analito medido en la muestra fortificada; CU es la concentración de analito medido en la muestra sin fortificar; CA es la concentración de analito agregado (valor nominal o teórico) en la muestra fortificada.
[AOAC-PVMC]
6.1.34 REPETIBILIDAD: Es la precisión bajo las condiciones de repetibilidad, es decir, condiciones donde los resultados de análisis independientes se obtienen con el mismo método en ítems de análisis idénticos en el mismo laboratorio por el mismo operador utilizando el mismo equipamiento dentro de intervalos cortos de tiempo.
[ISO 3534-1]
6.1.35 REPETIBILIDAD (DE RESULTADOS DE MEDIDAS): Es la proximidad entre los resultados de las medidas sucesivas del mismo analizado llevado a cabo en las mismas condiciones de medidas.
[ISO Orange Book]
6.1.36 REPETIBILIDAD (DE UN INSTRUMENTO DE MEDICION): Es la capacidad que tiene un instrumento de medición para brindar señales similares cercanas a las aplicaciones repetidas del mismo analizado bajo las mismas condiciones de medidas.
[ISO Orange Book)
6.1.37 REPRODUCIBILIDAD: Es la precisión bajo las condiciones de reproducibilidad, es decir, condiciones donde los resultados de los análisis se obtienen con el mismo método en ítems idénticos de análisis en distintos laboratorios con diferentes operadores usando distintos equipos.
Nota: Una declaración válida de reproducibilidad requiere especificación de las condiciones cambiadas.
La reproducibilidad se puede expresar en forma cuantitativa en términos de dispersión de los resultados.
[ISO 3534-1]
6.1.38 RESULTADO DE UNA MEDIDA: Es el valor atribuido al analizado, obtenido por medida.
Nota: Cuando se utiliza el término resultado de una medida, debe ser claro si se refiere a resultado no corregido, resultado corregido y si se promediaron varios valores. Una declaración completa del resultado de una medida incluye información acerca de la incertidumbre de medida.
[VIM 1993]
6.1.39 ROBUSTEZ (ROBUSTNESS): La robustez de un procedimiento analítico es una medida de su capacidad para permanecer inalterado por variaciones pequeñas pero deliberadas en parámetros del método y brinda una indicación de su confiabilidad durante el uso normal.
[ICH Q2A, CPMP/ICH/ 381/95]
6.1.40 SELECTIVIDAD (O ESPECIFICIDAD): Es la capacidad que tiene el método para determinar exacto y específicamente el analito de interés en la presencia de otros componentes en una matriz de muestra bajo condiciones del análisis establecidas.
[NATA Tech Note # 13]
6.1.41 SELECTIVIDAD (EN ANALISIS): Cualitativa: el grado en que otras sustancias interfieren con la determinación de una sustancia de acuerdo con un procedimiento dado.
[IUPAC Compendium of Chemical Technology, 1987]
6.1.42 SENSIBILIDAD: Es el cambio en la respuesta del instrumento de medición dividido por el cambio correspondiente en el estímulo.
Nota: El estímulo, por ejemplo, puede ser cantidad de analizado presente. La sensibilidad puede depender del valor del estímulo. A pesar de que esta definición se aplica claramente a un instrumento de medición, también se puede aplicar al método analítico globalmente, teniendo en cuenta otros factores tales como el efecto de los pasos de concentración.
[VIM 1984 y IUPAC Orange Book]
6.1.43 TRAZABILIDAD: Es la propiedad de un resultado de una medida o valor de un estándar por el cual se puede relacionar, con la incertidumbre establecida, a referencias establecidas, usualmente normas nacionales e internacionales (es decir, mediante una cadena ininterrumpida de comparaciones).
Nota: Los estándares a los que se refieren aquí son los estándares de medidas más que los estándares escritos (normas).
[ISO/IEC Guide 30-1992, 3.8]
6.1.44 UMBRAL DE DISCRIMINACION: Es el cambio más pequeño en un estímulo que produce un cambio perceptible en la respuesta de un instrumento de medición.
Nota: El umbral de discriminación puede depender, por ejemplo, del ruido (interno o externo), fricción, amortiguamiento, inercia, cuantificación.
[VIM 1984]
6.1.45 VALIDACION: Confirmar por medio de examen y provisión de evidencia objetiva que se cumplen los requisitos particulares para un uso propuesto específico.
[ISO 8402:1994]
6.1.46 VALIDACION DEL METODO:
6.1.46.1. Es el proceso para establecer las características de performance y limitaciones del método y la identificación de influencias que pueden cambiar estas características y hasta que punto.
¿Que analitos se pueden determinar en qué matrices en la presencia de qué interferencias?
Dentro de esas condiciones ¿qué niveles de precisión y exactitud se pueden lograr?
6.1.46.2. Es el proceso para verificar que un método es apto para ese propósito, es decir, para usarlo para resolver un problema analítico particular.
Nota: El punto 6.1.46.1. es aplicable cuando el método se desarrolla sin un problema particular previsto. El punto 6.1.46.2. es aplicable cuando el método se desarrolla con un fin específico. En química analítica el otro uso del término de validación es en el contexto de instrumental. La validación de instrumental se usa para describir el proceso de establecer que un instrumento en un momento dado se pueda operar de acuerdo con la especificación de diseño. Este proceso se podría lograr, por ejemplo, por medio de la calibración o control de performance.
6.1.47 VALOR: Valor de referencia aceptado.
Un valor que sirve como referencia de comparación previamente acordada y el cual deriva de:
A. Un valor establecido o teórico, basado en principios científicos.
B. Un valor asignado o certificado, basado en el trabajo experimental de algunas organizaciones nacionales e internacionales.
C. Un valor consensuado o certificado, basado en el trabajo experimental colaborativo bajo el auspicio de un grupo científico o de ingeniería.
D. Cuando a), b) y c) no están disponibles, la experimentación de una cantidad (mensurable), es decir, la medida de una población especificada de medidas.
[ISO 3534-1]
6.1.48 VALOR VERDADERO: Es el valor consistente con la definición de una cantidad particular dada.
[VIM 1993]
6.1.49 VALOR VERDADERO CONVENCIONAL: Valor atribuido a la cantidad particular y aceptada, algunas veces por convención, que tiene una incertidumbre adecuada para el propósito dado.
Nota: El valor verdadero convencional algunas veces se llama valor asignado, la mejor estimación del valor, valor convencional o valor de referencia. Frecuentemente, se usa una serie de resultados de medidas de una cantidad para establecer un valor verdadero convencional.
[VIM 1993]
6.1.50 VERACIDAD: Es el Grado de concordancia entre el valor promedio obtenido de una gran serie de resultados de análisis y un valor de referencia aceptado.
Nota: La medida de veracidad normalmente se expresa en términos de desviación. Generalmente, no se recomienda tomar a la veracidad como referencia para evaluar la exactitud del valor de la medida.
[ISO 3534-1]
6.1.51 VERIFICACION: Confirmar por medio de examen y aporte de evidencia objetiva que se cumplen los requisitos específicos.
[VIM 1993]
6.2 DISEÑO, EJECUCION E INFORME: El profesional responsable de la validación interna del método analítico en cuestión deberá diseñar el protocolo de validación, describiéndolo por escrito, paso a paso en forma de instrucción abarcando como mínimo los siguientes aspectos.
6.2.1 DISEÑO: Definición de la aplicación, propósito y alcance del método (considerando compuestos, matrices, tipo de información: cualitativa o cuantitativa, límites de detección y cuantificación, rango lineal, precisión y exactitud, etc.).
Definición de los parámetros de rendimiento y criterio de aceptación (deberá respetarse en el proceso de diseño los criterios que para cada tipo de análisis se describen en la presente instrucción).
Diseño de los experimentos de validación.
Descripción de las características relevantes requeridas para el equipamiento.
Calificación de los materiales, estándares y reactivos.
El protocolo de validación deberá ser evaluado y autorizado para su ejecución por el Coordinador del Area o Jefe del Departamento involucrado.
6.2.2 EJECUCION: Realización de los experimentos de pre-validación.
Ajuste de los parámetros del método y/o los criterios de aceptación si fuera necesario.
Realización de los experimentos de validación interna en forma completa.
Desarrollo los Procedimientos Operativos (POS) para ejecutar el método en la rutina.
Definición del tipo y frecuencia de pruebas adecuadas para el sistema y/o control de calidad analítico (CCA) para controles de rutina.
6.2.3 INFORME: El profesional responsable deberá documentar los experimentos y resultados de la validación en el informe respetando el siguiente esquema:
El objetivo y alcance del método (aplicabilidad, tipo).
Tipo de compuestos y de matriz.
Detalle de drogas, reactivos, estándares de referencia, y preparación de muestras de control.
Lista de equipos y requisitos de funcionamientos y rendimiento.
Procedimientos para los controles de calidad de estándares y drogas usadas.
Parámetros del método.
Parámetros críticos determinados para la comprobación de robustez (cuando corresponda).
Detalle de condiciones e implementación de los experimentos incluyendo la preparación de la muestra.
Procedimientos estadísticos y cálculos representativos.
Procedimientos para el control de calidad en la rutina.
Gráficos representativos, cromatogramas, espectros y curvas de calibración.
Criterios aceptación de los datos.
Incertidumbre esperada para los resultados de la medida.
Criterio para la revalidación.
Consideraciones seguridad.
Persona que desarrolló e inicialmente validó el método.
Resumen y conclusiones.
El Coordinador del área/Jefe de Departamento evaluará y aprobará el informe presentado, de cumplir con los criterios establecidos el método será incluido en el MANUAL DE METODOS ANALITICOS del área.
6.3 PARAMETROS A SER EVALUADOS SEGUN LA CATEGORIA DE METODO
|
PARAMETROS |
METODO CAT. I |
METODO CAT. II |
METODO CAT. III |
|
|
Cuantitativo |
Test Límite |
|||
|
Estabilidad |
Si |
Si |
Si |
* |
|
Selectividad |
Si |
Si |
Si |
Si |
|
Linealidad |
Si |
Si |
No |
* |
|
Rango |
Si |
Si |
* |
* |
|
Exactitud y Recuperación |
Si |
Si |
No |
* |
|
Precisión |
Si |
Si |
* |
Si |
|
MND |
No |
Si |
Si |
* |
|
MNC |
No |
Si |
Si |
* |
|
Estudio comparativo |
Si * |
* |
* |
|
|
Robustez |
Si |
Si |
Si |
Si |
(Punto 6.3 sustituido por art. 5° de la Disposición N° 1/2002 Laboratorios y Control Técnico del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O.30/7/2002. Vigencia: a partir de su publicación en B.O.)
En casos específicos de análisis puede ser necesario.
Categoría I: Métodos Analíticos para la cuantificación de materia prima o principio activo en producto terminado.
Categoría II: Métodos Analíticos para determinar impurezas en materia prima o compuestos de degradación en producto terminado. También para análisis de residuos en material biológico o alimentos.
Categoría III: Métodos Analíticos para determinar las características de funcionamiento como disolución o liberación de droga.
6.4 REQUISITOS A CUMPLIR EN EL PROCESO DE VALIDACION:
6.4.1 Linealidad y Sensibilidad con Patrones
6.4.1.1 Antes de la preparación de los patrones se efectuará el control de pureza de los solventes correspondientes, cuando se considere necesario.
6.4.1.2 Se deben analizar todos los patrones convenientemente agrupados.
6.4.1.3 Para estudiar la linealidad y sensibilidad de respuesta de los respectivos detectores se trabajará de la siguiente forma, donde CT es la concentración de trabajo:
6.4.1.3.1 Cromatografía, Gaseosa, Espectrofotometría de Absorción Atómica, Cromatografía sobre placa delgada (TLC/HPTLC) y HPLC: Blanco y CINCO (5) niveles de concentración: 0.25 CT, 0.5 CT, 0.75 CT, 1 CT y 2 CT, efectuar inyecciones por triplicado.
NOTA: Para detector selectivo de masa 0.5 CT, 1 CT y 2 CT, efectuar inyecciones por triplicado.
Se deben seleccionar por lo menos TRES (3) iones para la identificación y cuantificación. Determinar el tiempo de retención relativo al standard interno. El intervalo de aceptación es: ± 0.5%
Se grafica respuesta (área o altura) relativa al standard interno en función de masa de analito inyectada. Establecer la relación de abundancias para los iones seleccionados con relación al ion base.
6.4.1.3.2 Inmunoanálisis (IA) RIA o ELISA:
RIA: Blanco y 6 niveles de concentración: entre DIEZ (10) y UN MIL (1000) pg para todas las sustancias (excepto estradiol), por duplicado. Estradiol: entre DOS (2) y DOSCIENTOS (200) pg, por duplicado.
ELISA: Blanco y SEIS (6) niveles de concentración: entre CUATRO (4) y CUATROCIENTOS (400) pg para todas las sustancias, por duplicado.
6.4.1.4 Se grafica respuesta (área, altura o relación de áreas/alturas en el caso de utilizar standard interno) en función de la masa de analito inyectada y se efectúa el tratamiento estadístico de los datos, determinado ordenada al origen, pendiente (sensibilidad), coeficiente de correlación y coeficiente de variación porcentual para cada nivel de concentración.
NOTA: En Inmunoanálisis, cuando existe mucha dispersión entre los valores experimentales, puede ser eliminado un máximo de DOS (2) puntos siempre que no pertenezcan al mismo nivel de concentración.
En el caso de inyección manual el coeficiente de variación porcentual (CV%) debe ser menor que CINCO POR CIENTO (5%) para cada nivel de concentración. Cuando se utiliza inyector automático el CV% debe ser menor que DOS POR CIENTO (2%), excepto para HPLC y GC-MS CUATRO POR CIENTO (4%). El coeficiente de correlación no debe ser inferior a 0.990 excepto 0.980 en inmunoanálisis.
6.4.1.5 HPLC preparativo para Confirmación por IS/GC-MS:
A. Determinar por HPLC el tiempo de retención del analito problema, utilizando un patrón con el analito de interés y todos aquellos que den reacción inmunológica cruzada con el mismo, a una concentración suficiente para ser detectados y asegurar la separación cromatográfica.
B. Inyectar Blanco de reactivos y Patrón en una concentración igual a la CT o al Nivel de Acción, por triplicado y aislar las fracciones correspondientes al tiempo de retención que se determinó mediante el procedimiento indicado en A.
C. Efectuar el análisis por IA o GC-MS a dichas fracciones.
D. Determinar Recuperación% y CV%.
6.4.2 Precisión, Repetibilidad, Reproducibilidad y Selectividad con Muestras Fortificadas.
Se deberán analizar nuestras fortificadas para todos los analitos en diferentes niveles de concentración según el método analítico que se aplique. Donde CNR es la concentración nominal de referencia y está relacionada con el Límite Máximo de Residuos (LMR) o el Nivel de Acción (NA).
6.4.2.1 Cromatografía Gaseosa, Espectrofotometría de Absorción Atómica y Cromatografía sobre placa delgada (TLC/HPTLC) y HPLC: Procesar Blanco de matriz y muestras fortificadas a CINCO (5) niveles de concentración: 0.25 CNR, 0.5 CNR, 0.75 CNR, 1 CNR y 2 CNR por quintuplicado, distribuidos en días distintos (las muestras utilizadas para esta evaluación deberán proceder de distintos animales o lotes a efectos de contemplar la variabilidad entre muestras).
Cromatografía gaseosa con detector selectivo de masas (GC-MS): Procesar Blanco de matriz y muestras fortificadas a TRES (3) niveles de concentración: 0.5 CNR, 1 CNR y 2 CNR por quintuplicado, distribuidos en días distintos (las muestras utilizadas para esta evaluación deberán proceder de distintos animales o lotes a efectos de contemplar la variabilidad entre muestras).
Verificar que la relación de abundancias para los iones seleccionados se halle comprendida entre el valor promedio obtenido con los patrones ± 20%, (para relaciones de abundancia menores que 50%) y ± 10% (para relaciones de abundancia mayores que 50%).
El intervalo debe ser determinado en forma relativa respecto al valor de la relación promedio obtenido.
De acuerdo a la sensibilidad obtenida para técnica, deberá definirse el valor mínimo de concentración que podrá ser identificada con certeza (Mínimo Nivel de Confirmación). Para los residuos de anabólicos en orina, este valor deberá corresponder al Nivel de Acción respectivo establecido en base al Background para RIA.
Procesar blanco de matriz por cuadruplicado y muestras fortificadas a ese nivel de concentración por sextuplicado, distribuidos en días diferentes según se indicó anteriormente. Verificar el cumplimiento de las relaciones de abundancias.
6.4.2.2 Inmunoanálisis: Procesar Blanco de reactivos y muestras fortificadas a CUATRO (4) niveles de concentración: 0.5 CNR, 0.75 CNR, 1 CNR y 2 CNR por sextuplicado y VEINTE (20) Muestras Blanco distribuidos en días distintos.
NOTA: Los resultados obtenidos a 0.25 CNR se podrían descartar del análisis estadístico si su CV es mayor al aceptable, pudiendo ser utilizados en la determinación del MNC. Los valores hallados no deben ser corregidos por recuperación, excepto para técnicas de inmunoanálisis en tejidos donde deben corregirse por recuperación extractiva de cada muestra, calculada por el agregado de hormona marcada.
6.4.2.3 Se determina Recuperación Porcentual (R%) para cada fortificado y CV% para cada nivel de concentración. Se grafica Concentración Hallada en función de Concentración Nominal. Se efectúa el tratamiento estadístico de los datos, determinando ordenada al origen pendiente (recuperación promedio) y coeficiente de correlación.

MINIMO NIVEL DETECTABLE (MND).
1) Es la concentración nominal determinada a partir de la ordenada al origen de la hipérbola superior de confianza para un nivel de significación de 0.05.
Cuando se considere necesario los valores obtenidos para MND deberán ser confirmados experimentalmente, fortificando muestras de la misma matriz a los niveles de concentración respectivos y efectuando su análisis con el mismo número de replicados que en la validación correspondiente.
2) Inmunoanálisis: se determina como el Promedio + 3 Desvíos Standard del análisis de VEINTE (20) muestras blanco.
MINIMO NIVEL CUANTIFICABLE (MNC).
A. Se grafica %CV para cada nivel de concentración y se determina como la concentración nominal para la cual el %CV es:
A1- DIEZ POR CIENTO (10%) para concentraciones mayores que 100 ng/g.
A2- VEINTE POR CIENTO (20%) para concentraciones menores o iguales que 100 ng/g.
(Subítem A sustituido por art. 3° de la Disposición N° 6/2004 de la Dirección de Laboratorios y Control Técnico B.O. 30/11/2004. Vigencia: a partir de su publicación en el Boletín Oficial).
B. Inmunoanálisis: Se determina como el Promedio + 6 Desvíos Standard del análisis de VEINTE (20) muestras blanco.
6.4.2.7 HPLC preparativo para Confirmación por IA/GC-MS:
A. Procesar Blanco de matriz y Fortificados en una Concentración igual a la CNR o al Nivel de Acción (NA) por cuadruplicado y aislar las fracciones correspondientes al tiempo de retención que se determinó mediante el procedimiento indicado en 6.4.1.5 A.
B. Efectuar a dichas fracciones el análisis por IA o GC-MS.
C. Determinar Recuperación% y CV%.
6.4.3 CALCULO DE INCERTIDUMBRE
Los laboratorios deben tener y aplicar procedimientos para estimar la incertidumbre de medición, deben tratar de identificar todos los componentes de la incertidumbre, entre ellas pueden encontrarse: definición incompleta del mensurando, definición imperfecta del mensurando, muestreo, preparación de muestras, condiciones ambientales, desviaciones personales en la lectura de instrumentos analógicos, límites en la discriminación o resolución del instrumento, valores inexactos de los patrones y materiales de referencia utilizados, valores inexactos de constantes y otros parámetros obtenidos de fuentes externas y utilizados en el algoritmo para la obtención de datos.
Deben procesarse como mínimo VEINTICINCO (25) fortificados correspondientes a CINCO (5) niveles de concentración por quintuplicado, por lo tanto se dispone de VEINTICINCO (25) valores de Recuperación Porcentual (R %).
Calcular la desviación standard relativa DSR, según:
|
|
DS (R %) |
|
DSR = |
––––––––––––– |
|
|
R % (Prom.) |
DS (R %): desvío standard de las R %.
R % (Prom.): recuperación promedio.
Calcular la Incertidumbre expandida (U) para una medida "c" multiplicando la DSR por un factor de cobertura "k" y c.
U = DSR x k x c
El valor de k es 2 para un nivel de confianza de 95%.
La Incertidumbre Porcentual también puede ser determinada a partir de por lo menos VEINTICINCO (25) valores experimentales de R % correspondientes a análisis de rutina, según el mismo procedimiento.
En este caso es recomendable disponer de datos de R% para distintos niveles de concentración dentro del rango analítico. En las distintas tandas de muestras analizadas deberían procesarse recuperados a diferentes concentraciones dentro del rango analítico y recalcular la Incertidumbre cuando se disponga de por lo menos VEINTICINCO (25) valores de R % para verificar la validez del valor de Incertidumbre determinado anteriormente.
Se recomienda trabajar con CINCUENTA (50) datos, por lo tanto cuando se cuente con CINCUENTA (50) valores de R% corresponde recalcular la incertidumbre.
La incertidumbre debe ser recalculada anualmente o cuando se produce un cambio importante en el método, por ejemplo adopción de nuevas metodologías, cambio del personal, nuevos equipos (por ejemplo: detector, espectrómetro, columna analítica) que muestren respuesta del analito marcadamente diferente.
EVALUACION DE LA ROBUSTEZ
Se utiliza el procedimiento de Youden y Steiner, que permite evaluar siete variables con el análisis de sólo ocho muestras.
Las variables deben ser elegidas estratégicamente. Se examina el método escrito y se identifican aquellas etapas que posiblemente pueden afectar los resultados finales, además de otras variables "habituales" como la concentración de reactivos, temperatura, tipo y tiempo de agitación, tipo de filtro, etc.
Cada variable se estudia mediante un valor (o cualidad cuando esto no es posible) alto (A, B,...G) y otro bajo (a, b,...g) y se diseñan ocho pruebas según el ejemplo de la Tabla 1 (la que consta a posteriori). Los resultados se representan con letras desde s hasta z.
A partir de los resultados puede calcularse el efecto de cada una de las variables haciendo la media de los cuatro análisis que contienen la variable en su valor más alto (mayúsculas) y aquellos que corresponden al valor más bajo (minúsculas). Así, el efecto de cambio del Factor "A" a "a" se mide por la diferencia:
(s + t + u + v)/4 - (w +x +y + z)/4
Es decir, la media de los resultados (s+t+u+v) equivale a "A" porque las SEIS (6) variables restantes presentes en estos cuatro resultados se anulan entre sí como consecuencia de que existen siempre DOS (2) mayúsculas y DOS (2) minúsculas de cada variable. Análogamente, la media de los resultados (w + x + y + z) equivale a "a".
Se calcula el efecto de cada uno de los factores. Finalmente el efecto de cambio de "G" a "g" se mide por la diferencia (s + v + x + y)/4 - (t + u + w + z)/4.
Al comparar los dos valores medios se conoce la influencia de la variable en el estudio. Para cualquier otra variable se puede proceder de manera similar.
Tabla 1: Test de Robustez de Youden para un método analítico
|
VALOR DE LAS VARIABLES |
ANALISIS |
|||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
|
A, a |
A |
A |
A |
A |
a |
a |
a |
a |
|
B, b |
B |
B |
b |
b |
B |
B |
b |
b |
|
C, c |
C |
c |
C |
c |
C |
c |
C |
c |
|
D, d |
D |
D |
d |
d |
d |
d |
D |
D |
|
E, e |
E |
e |
E |
e |
e |
E |
e |
E |
|
F, f |
F |
f |
f |
F |
F |
f |
f |
F |
|
G, g |
G |
g |
g |
G |
g |
G |
G |
g |
|
Resultados |
s |
t |
u |
v |
w |
x |
y |
z |
Estableciendo las siete comparaciones posibles (A-a,...G-g) puede conocerse el efecto de cada variable; cuanto mayor sea la diferencia, mayor influencia tendrá dicha variable en el método analítico. Si cualquiera de estas diferencias entre los promedios de subgrupos de cuatro es mayor que (2)1/2 DS entre los replicados llevados a cabo en las mismas condiciones, es indicación de que el método es sensible a los cambios del factor involucrado. Estas variables recibirán especial atención al redactar el método, remarcando la necesidad de un estricto control para obtener resultados de calidad.
Nota 1: Los factores a estudiar no deben ser necesariamente siete; puede considerarse un número menor de variables. Esto no afectará el balance del diseño del experimento siempre que se lleven a cabo los ocho ensayos indicados.
Nota 2: una información adicional de este test de Youden es que la desviación standard de los resultados s a z constituye una medida excelente de la imprecisión previsible del método cuando se utiliza para el análisis de rutina, ya que este procedimiento introduce deliberadamente el tipo de variación en las variables que puede esperarse que ocurra durante el empleo normal del método.
CRITERIOS PARA EL CALCULO DE CCµ Y CCb.
Grupo A: Sustancias que poseen efectos anabólicos o sustancias no autorizadas.
Grupo B: Medicamentos veterinarios y contaminantes.
Límite máximo de residuo (MRL): concentración máxima de residuo legalmente permitida o aceptada en alimentos bajo leyes internacionales.
LIMITE MINIMO DE FUNCIONAMIENTO REQUERIDO (MRPL): Mínimo contenido de analito en una muestra que debe ser detectado y confirmado por un laboratorio de control de residuos.
LIMITE DE DECISION (CCµ)
Límite en el cual y a partir del cual se puede concluir con una probabilidad de error a que la muestra no es conforme (es positiva).
CAPACIDAD DE DETECCION (CCb)
Contenido mínimo de la sustancia que puede ser detectado, identificado y/o cuantificado en una muestra con una probabilidad de error b.
METODOS DE SCREENING SUSTANCIAS PROHIBIDAS
LIMITE DE DECISION (CCµ): para anabólicos por RIA es igual al obtenido en el estudio de background realizado en base a los datos de todos los laboratorios.
CAPACIDAD DE DETECCION (CCb): tomar VEINTE (20) muestras blanco, fortificar al nivel del Límite de Decisión y analizar. Es igual al Límite de decisión +1.64 DS.
METODOS DE CONFIRMACION SUSTANCIAS PROHIBIDAS
LIMITE DE DECISION (CCµ): es la concentración nominal correspondiente a la ordenada al origen + 2.33 DS de la reproducibilidad intralaboratorio de los datos de validación (equivalente a la ordenada al origen de la hipérbola superior para un nivel de confianza del 99%).
CAPACIDAD DE DETECCION (CCb): es igual al Límite de decisión + 1.64 DS.
METODOS DE CONFIRMACION SUSTANCIAS PERMITIDAS
LIMITE DE DECISION (CCµ): en aquellos casos que la CNR (Concentración Nominal de Referencia), que está relacionada con el LMR, de la validación es igual al LMR o el LMR está incluido en la curva, se trabaja con los datos de validación y es igual al LMR + 1.64 DS (al nivel del LMR).
CAPACIDAD DE DETECCION (CCb): es igual al Límite de decisión + 1.64 DS (al nivel del LMR).
(Item 6.4.3 incorporado por art. 5° de la Disposición N° 6/2004 de la Dirección de Laboratorios y Control Técnico B.O. 30/11/2004. Vigencia: a partir de su publicación en el Boletín Oficial).
7. REGISTRO Y ARCHIVO:
Conservar los registros como mínimo durante SEIS (6) años, en forma permanente mientras esté en uso.
(Item 7 sustituido por art. 4° de la Disposición N° 6/2004 de la Dirección de Laboratorios y Control Técnico B.O. 30/11/2004. Vigencia: a partir de su publicación en el Boletín Oficial).
8. ANEXO:
Anexo I: Distribución de Copias.
9. HISTORICO:
No contiene.
10. REFERENCIAS:
AOACI Peer Verified Methods 1999, Eurachem Guide 1998, Resolución RC 300/99 y 825/99 Directivas de la Coordinación de Residuos Químicos.
LISTA DE DISTRIBUCION
|
NOMBRE |
TIPO DE COPIA |
FIRMA |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
..................................................................
Firma Responsable – Aclaración
ANEXO V
INFORME DE RESULTADOS
1. OBJETO:
Establecer el criterio para el informe de resultados.
2. ALCANCE:
Debe aplicarse a muestras de carácter oficial. Será adaptado por cada Laboratorio de la Red, según su organigrama.
3. AREAS AFECTADAS:
Serán adaptadas por cada Laboratorio de la Red, según su organigrama.
4. RESPONSABILIDADES:
Profesionales y técnicos. Serán adaptadas por cada Laboratorio de la Red, según su organigrama.
5. RELACIONES:
Serán adaptadas por cada Laboratorio de la Red, según su organigrama.
6. DESARROLLO
6.1 RESUMEN:
Se establece la metodología para confirmar presuntos positivos de residuos de medicamentos, contaminantes y anabólicos, y los criterios para el informe de resultados.
6.2 METODOS RECONOCIDOS PARA CONFIRMACION:
6.2.1 Cuando se utiliza IA (RIA Y ELISA) o HPTLC como métodos de criba (screening) podrán emplearse como métodos de confirmación los siguientes:
6.2.1.1 Cromatografía líquida de alta presión para la separación y purificación del analito seguida del método de inmunoensayo de la fracción (HPLC-IA).
6.2.1.2 Cromatografía gaseosa con detector selectivo de masas de la fracción (HPLC/GC-MS).
Cuando la concentración de la sustancia, que ha dado positivo por el método de Inmunoanálisis utilizado en el Screening, no sea suficiente como para ser detectada por el detector del cromatógrafo líquido, deberá procederse de la siguiente forma:
A- Se determinará por HPLC el tiempo de retención del analito problema, utilizando un patrón que contenga el analito de interés y aquellos que den reacción imnunológica cruzada, a una concentración suficiente para ser detectados.
B- Se inyectará la muestra problema en iguales condiciones de trabajo y se aislará/n la/s fracción/ es correspondiente/s al/los tiempos de retención que se determinó/aron mediante el procedimiento indicado en A.
Conservar registros cromatográficos de A y B.
C- A dicha fracción se le efectuará el análisis por IA o GC-MS.
6.2.2 Cromatografía gaseosa con detector selectivo de masas (GC-MS).
6.2.3 Cromatografía Líquida (HPLC)
Cuando se utiliza HPLC como método de criba (screening), para todos los presuntos positivos de analitos detectados en concentraciones mayores al MND (antiparasitarios, nitrofuranos, sulfonamidas, etc.) con el detector de arreglo de diodos (DAD) y/o el detector de fluorescencia (FLD), se debe comparar el espectro obtenido del presunto positivo con el del patrón correspondiente. En el caso de utilizar para el análisis de screening un detector de fluorescencia que no permita la toma de espectros, la confirmación de un presunto positivo debe realizarse con el detector DAD y comparar el espectro con el del patrón respectivo y el del pico correspondiente en una muestra fortificada.
6.2.4 Cromatografía Gaseosa (GC)
Cuando se utiliza GC como método de criba (screening), los presuntos positivos de analitos identificados en concentraciones mayores que el Mínimo Nivel Detectable con detector específico para su estructura molecular, pueden ser confirmados por el mismo método utilizando dos o más columnas de distinta polaridad o por otra técnica diferente aplicada para su análisis (por ejemplo, Cloranfenicol que puede determinarse también por 1 método de RIA, y confirmarse por HPLC-RIA, HPLC-DAD o GC-MS).
En todos los casos de presuntos positivos las muestras deben ser analizadas por duplicado. Los registros de las confirmaciones deben conservarse por tres (3) años y el laboratorio debe conservar las contramuestras perfectamente identificadas bajo condiciones de seguridad (en custodia) seis (6) meses.
6.3 PROTOCOLOS DE ANALISIS:
6.3.1 Cuando se informan los valores numéricos de concentración en el protocolo, los mismos deben estar corregidos por la correspondiente recuperación. Para Residuos de Plaguicidas, PCB’s y Elementos químicos, cuando el método tenga una recuperación entre 80 y 110% no debe ser corregido por recuperación.
6.3.2 Informar la recuperación de la tanda.
6.3.3 Informar la Incertidumbre
Deberá informarse la incertidumbre calculada siguiendo las exigencias de la normativa vigente y aplicable en la materia, cuando la autoridad regulatoria o el cliente lo solicite.
(Subítem 6.3.3 sustituido por art. 6° de la Disposición N° 6/2004 de la Dirección de Laboratorios y Control Técnico B.O. 30/11/2004. Vigencia: a partir de su publicación en el Boletín Oficial).

6.3.5 Protocolo básico:
La expresión de los resultados deberá seguir los criterios detallados a continuación:
ND: cuando el valor hallado es menor que el Mínimo Nivel Detectable. Según AOACI Peer Verified Methods cuando la recuperación es < que 100%, el valor obtenido se debe corregir por recuperación antes de decir que es menor que el MND.
En inmunoanálisis ND: cuando el valor hallado es menor que el Nivel de Acción.
<MNC: cuando el valor hallado está comprendido entre el MND y el MNC, siendo MNC el valor numérico del Mínimo Nivel Cuantificable para ese analito, excepto para inmunoanálisis.
VALOR HALLADO: cuando la concentración hallada es mayor que el MNC. Deberá estar corregido por la recuperación respectiva en los casos que corresponda.
En todos los casos se indicará las unidades que se utilizan:
ppm (mg/Kg o Litro)
ppb (microg/Kg o Litro)
ppt (nanog/Kg o Litro)
Para los rubros con programas de muestreo dirigido respetar los períodos en vigencia, mientras que para los rubros de muestreo insesgado, las muestras deberán informarse dentro de los 30 días corridos. Res RS 813/99.
7. REGISTRO Y ARCHIVO:
Conservar los registros como mínimo durante SEIS (6) años.
(Item 7 sustituido por art. 7° de la Disposición N° 6/2004 de la Dirección de Laboratorios y Control Técnico B.O. 30/11/2004. Vigencia: a partir de su publicación en el Boletín Oficial).
8. ANEXO:
Anexo 1: Distribución de Copias.
Anexo 2: Modelo de Protocolo.
9. HISTORICO:
No contiene.
10. REFERENCIAS:
AOACI Peer Verified Methods 1999, Eurachem Guide 1998, Resolución RS 813/99, Resolución RC 300/99 y 825/99, Codex Alimentarius 2001 y Directivas de la Coordinación de Residuos Químicos.