
Secretaría de Políticas, Regulación y Relaciones Sanitarias y Secretaría de Agricultura, Ganadería, Pesca y Alimentos
CODIGO ALIMENTARIO ARGENTINO
Resolución Conjunta 71/2002 y 390/2002
Modifícanse artículos del mismo, con la finalidad de adecuar la definición del aceite de oliva a las normas internacionales.
Bs. As., 27/12/2002
VISTO la Ley 18284, los Artículos 535, 536 y 1414 del Código Alimentario Argentino y el Expediente N° 1-47-2110-1108-02-7 del Registro de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica y,
CONSIDERANDO:
Que es necesaria la actualización de los parámetros de genuinidad del aceite de oliva y del aceite de orujo de aceituna que figuran en el Código Alimentario Argentino.
Que existen antecedentes internacionales tales como, la Norma del Codex para los aceites de oliva vírgenes y refinados y para los aceites refinados de orujo de aceituna STAN 33; Alinorm 01/17 de la Comisión del Codex Alimentarius; Reglamento CE N° 2472/97 de la Unión Europea; Resolución ANVISA 482/99 de Brasil; Norma COI T.20 Documento N° 17 y 10.
Que se considera necesaria la incorporación de nuevos parámetros de genuinidad para el aceite de oliva y para el aceite de orujo de aceituna, conservando las especificaciones ya establecidas en los artículos 535 y 536 del Código Alimentario Argentino.
Que al incluir parámetros nuevos de genuinidad, se hace necesaria la incorporación al Código Alimentario Argentino de las técnicas de determinación de los mismos.
Que en virtud de lo expuesto resulta necesario modificar los artículos 535, 536 y 1414 del mencionado Código.
Que es necesario modificar la denominación del aceite de oliva con el fin de adecuarla a las normas internacionales.
Que la Comisión Nacional de Alimentos ha tomado intervención expidiéndose en tal sentido.
Que los Servicios Jurídicos Permanentes de los organismos involucrados han tomado la intervención de su competencia.
Que se actúa en virtud de las facultades conferidas por el Decreto N° 815/99).
Por ello,
EL SECRETARIO DE POLITICAS, REGULACION
Y
RELACIONES SANITARIAS Y EL SECRETARIO DE AGRICULTURA, GANADERIA, PESCA Y ALIMENTOS
RESUELVEN:
Artículo 1° — Sustitúyese el artículo 535 del Código Alimentario Argentino, el que quedará redactado de la siguiente forma:
"Artículo 535: Se denomina Aceite de oliva, al obtenido de los frutos de Olea europaea L.
Se denomina Aceite de oliva de presión, al obtenido fundamentalmente a partir del fruto entero y exclusivamente por procedimientos mecánicos y técnicos adecuados y purificado solamente por lavado, sedimentación, filtración y/o centrifugación (excluida la extracción por disolventes). Podrá designarse como Aceite de oliva virgen y se clasificará de acuerdo a su grado de acidez libre como: Clase Extra o Calidad Extra, Clase Fina o Calidad Fina y Clase Común o Calidad Común.
El aceite de oliva obtenido por presión y sometido a proceso de refinación se designará Aceite de oliva refinado o Aceite de oliva de presión refinado.
Con la designación de Aceite de Oliva (sin otra denominación) se entiende a una mezcla de aceite de oliva virgen con aceite de oliva refinado.
Sus características fisicoquímicas son:
Densidad relativa a 25/4°C: 0,9090 a 0,9130.
Indice de refracción a 25°C: 1,4665 a 1,4683.
Indice de yodo (Wijs): 79 a 89.
Indice de saponificación: 187 a 195.
Insaponificable, Máx.: 1,30%.
Indice de Reichert-Meissl, Máx: 1,2%.
Indice de Polenske, Máx.: 2,5%.
Indice de Bellier modificado (medio acético de precipit.), Máx.: 16° C.
Extinción específica:
- Aceite de oliva virgen a 232 y 270 nm., Máx.: 4,0 y 0,3 respectivamente.
- Aceite de oliva refinado a 270 nm: Máx 1,10 (variación máxima a cerca de 270 nm: 0,16).
- Aceite de oliva a 270 nm: Máx 0,90 (variación máxima acerca de 270 nm: 0,15). Acidez libre:
- Aceite de oliva virgen Clase Extra, Máx: 2,00 mg KOH/g (1,00% como ác. oleico).
- Aceite de oliva virgen Clase Fina, Máx: 4,00 mg KOH/g (2,00% como ác. oleico).
- Aceite de oliva virgen Clase Común, Máx: 6,60 mg KOH/g (3,30% como ác. oleico).
- Aceite de oliva Refinado, Máx: 0,60 mg KOH/g (0,30% como ác. oleico).
- Aceite de oliva, Máx 3,00 mg KOH/g (1,5% como ác. oleico).
Por Pérdida por calentamiento:
- Aceite de oliva virgen Clases Extra, Fino y Común, Máx: 0,15%.
- Aceite de oliva Refinado, Máx 0,05%.
- Aceite de oliva, Máx 0,10%.
Indice de peróxidos:
- Aceite de oliva virgen Clases Extra, Fino y Común, Máx: 30,0 miliequivalentes de Oxígeno por Kg.
- Aceite de oliva y Aceite de oliva Refinado: Máx 20,0 miliequivalentes de Oxígeno por kilogramo.
La composición de ácidos grasos determinada por cromatografía en fase gaseosa (ésteres metílicos por ciento) debe encuadrarse dentro de los siguientes límites:
- Acido láurico (C 12:0): No perceptible.
- Acido mirístico (C14:0): Menor de 0,1.
- Acido palmítico (C16:0): 7,5 a 20,00.
- Acido palmitoleico (C16:1): 0,3 a 3,5.
- Acido heptadecanoico (C17:0): Menor de 0,5.
- Acido heptadecenoico (C17:1): Menor de 0,6.
- Acido esteárico (C18:0): 0,5 a 5,0.
- Acido oleico (C18:1): 53,0 a 83,0.
- Acido linoleico (C18:2): 3,5 a 21,0.
- Acido linolénico (C18:3): Menor de 1,5.
- Acido araquídico (C20:0): Menor de 0,8.
- Acido behénico (C22:0): Menor de 0,2.
- Acido erúcico (C22: 1): No perceptible.
- Acido lignocérico (C24:0): Menor de 0,1.
El contenido de ácidos grasos trans (expresado como % respecto de los ácidos trasos totales), será el siugiente:
- Transoleico (C18:1T):
- Aceites (de oliva virgen: Menor de 0,05.
- Aceite (de oliva: Menor de 0,20. -
- Translinoleico + Translinolenico (C18:2 T + C18:3 T):
- Aceites de oliva vírgenes: Menor de 0,05.
- Aceite de oliva: Menor de 0,30.
La composición de esteroles (expresado como % de desmetilesteroles respecto del total en esteroles), será la siguiente:
- Colesterol: Menor o igual de 0,5.
- Brassicasterol: Menor o igual de 0,1.
- Campesterol: Menor o igual de 4,0.
- Estigmasterol: Menor que campesterol.
- D-7-stigmastenol: Menor o igual de 0,5.
- b-sitosterol + D-5-avenasterol + D-5-23-estigmastadienol + clerosterol + sitostanol + D-5-24- estigmastadienol: Mayor o igual de 93,0".
Art. 2° — Sustitúyese el artículo 536 del Código Alimentario Argentino, el que quedará redactado de la siguiente forma:
"Artículo 536: Se denomina Aceite de orujo de aceituno refinado, al obtenido de orujos de aceitunas, por medio de los disolventes autorizados y que ha sido neutralizado, blanqueado, desodorizado y desmargarizado no pudiendo ser sometido a procesos de reesterificación.
Sus características fisicoquímicas son las indicadas en el Artículo 535 a excepción de:
Insaponificable, Máx: 2,10%.
Pérdida por calentamiento, Máx: 0,05%.
Indice de Bellier modificado (medio acético de precipit.): no aplicable.
Debe presentar opalescencia estable a temperatura superior a 50’C y luego floculación.
Acidez libre, Máx: 0,60 mg KOH/g (0,30% como ác. Oleico).
Indice de peróxido, Máx: 20,0 miliequiv. de Oxígeno/Kg.
Composición de esteroles (expresado como % de desmetilesteroles respecto del total en esteroles): las indicadas en el Artículo 535 a excepción de:
- Brassicasterol: Menor o igual de 0,2.
Contenido de ácidos grasos trans:
- Transoleico (C18:1 T): Menor de 0,2 % de los ácidos grasos totales.
- Translinoleico + Translinolenico (C 18:2 T + C 18:3 T): Menor de 0,3 % de los ácidos grasos totales."
Art. 3° — Inclúyese en el artículo 1413 del Código Alimentario Argentino, el apartado 11.22, el cual quedará redactado de la siguiente forma:
"11.22 - DETERMINACION DE LA COMPOSICION Y DEL CONTENIDO DE ESTEROLES MEDIANTE CROMATOGRAFÍA DE GASES CON COLUMNA CAPILAR
1. OBJETIVO
El presente método describe un procedimiento para la determinación del contenido total e individual de esteroles en las materias grasas.
2. PRINCIPIO
Saponificación de la materia grasa, a la que se habrá añadido a-colestanol como patrón interno, con una solución etanólica de hidróxido de potasio; a continuación extracción del insaponificable con éter etílico.
Separación de la fracción de esteroles del extracto insaponificable mediante cromatografía en placa de sílica gel básica; los esteroles recuperados del gel de sílica se transforman en trimetilsililéteres y se analizan mediante cromatografía de gases con columna capilar.
Las condiciones de trabajo y materiales detallados a continuación podrán adecuarse a las características de la columna, el cromatógrafo y la disponibilidad de insumos con que cuente el laboratorio.
3. MATERIAL
3.1. Matraz de 250 ml, provisto de refrigerante de reflujo con juntas esmeriladas.
3.2. Embudos de separación de 500 ml.
3.3. Matraces de 250 ml.
3.4. Equipo completo de cromatografía en capa fina, utilizando placas de vidrio de 20 x 20 cm.
3.5. Lámpara ultravioleta de una longitud de onda de 366 o 254 nm.
3.6. Microjeringa de 100 ul y 500 ul.
3.7. Embudo cilíndrico filtrante con filtro poroso G3 (porosidad 15-40 mm), de 2 cm de diámetro y 5 cm de altura, aproximadamente, con un dispositivo adecuado para la filtración en vacío y una junta esmerilada macho 12/21.
3.8. Matraz cónico para vacío de 50 ml, con junta esmerilada hembra 12/21 que acople con el embudo filtrante (3.7).
3.9. Probeta de 10 ml de fondo cónico con tapón hermético.
3.10. Equipo de cromatografía de gases que pueda funcionar con columna capilar, provisto de un sistema de división de flujo formado por:
3.10.1. Horno para la columna, que pueda mantener la temperatura deseada con precisión de +/- 1°C.
3.10.2. Inyector con elemento vaporizador de vidrio tratado con persilano.
3.10.3. Detector de ionización de llama y convertidor-amplificador.
3.10.4. Registrador-integrador que pueda funcionar con el convertidor-amplificador (3.10.3), con un tiempo de respuesta no superior a 1 segundo y con velocidad de papel variable.
3.11. Columna capilar de vidrio o sílice fundida, de 20 a 30 m de longitud y de 0,25 a 0,32 mm de diámetro interno, recubierta interiormente de líquido SE-52, SE-54 o equivalente, con un espesor uniforme que oscile entre 0,10 y 0,30 mm.
3.12. Microjeringa de 10 ml para cromatografía de gases, con aguja endurecida.
4. REACTIVOS
4.1. Hidróxido de potasio, solución etanólica 2N aproximadamente: disolver, enfriando al mismo tiempo, 130 g de hidróxido de potasio (valoración mínima del 85 %) en 200 ml de agua destilada y completar hasta un litro con etanol. Conservar la solución en botellas de vidrio oscuro bien cerradas.
4.2. Éter etílico de calidad para análisis.
4.3. Sulfato sódico anhidro de calidad para análisis.
4.4. Placas de vidrio recubiertas con sílica gel, sin indicador de fluorescencia, de 0,25 mm de espesor (disponibles en el comercio ya preparadas para el uso).
4.5. Hidróxido de potasio, solución etanólica 0,2N: disolver 13 g de hidróxido de potasio en 20 ml de agua destilada y completar hasta un litro con etanol.
4.6. Tolueno para cromatografía.
4.7. Acetona para cromatografía. (Véase 5.2.2).
4.8. Hexano para cromatografía. (Véase 5.2.2).
4.9. Éter etílico para cromatografía. (Véase 5.2.2).
4.10. Cloroformo de calidad para análisis.
4.11. Solución patrón para cromatografía en capa fina: colesterol o fitoesteroles, solución al 5 % en cloroformo.
4.12. Solución de 2,7-díclorofluoresceína al 0,2 % en etanol. Para hacerla ligeramente básica se añaden algunas gotas de solución alcohólica 2N de hidróxido de potasio.
4.13. Piridina anhidra para cromatografía.
4.14. Hexametildisilazano.
4.15. Trimetilclorosilano.
4.16. Solución problema de trimetilsililéteres de los esteroles: preparar en el momento del uso a partir de esteroles obtenidos de aceites que los contengan.
4.17. a-colestanol, disolución al 0,2 % (m/N) en cloroformo (patrón interno).
4.18. Gas portador: hidrógeno o helio de calidad para cromatografía de gases.
4.19. Gases auxiliares:
- hidrógeno o helio de calidad para cromatografía de gases.
- aire de calidad para cromatografía de gases.
5. PROCEDIMIENTO
5.1. Preparación del insaponificable.
5.1.1. Con la microjeringa de 500 ml introducir en el matraz de 250 ml un volumen de disolución de a-colestanol al 0,2 % en cloroformo (4.17) que contenga una cantidad de a-colestanol correspondiente al 10 % aproximadamente del contenido de esteroles en la alícuota de la muestra para la determinación. Por ejemplo, para 5 g de muestra añadir 500 ml de la solución de a-colestanol al 0,2 %, si se trata de aceite de oliva, y 1500 mI, si se trata de aceite de orujo de aceituna.
Evaporar en corriente de nitrógeno hasta sequedad y, a continuación, pesar con precisión, en el mismo matraz, 5 g de muestra seca y filtrada.
En el caso de aceites y grasas animales o vegetales con un alto contenido de colesterol puede producirse un pico cuyo tiempo de retención sea idéntico al del colestanol. En tal caso el análisis de la fracción de esteroles debe realizarse dos veces: con patrón interno y sin él.
5.1.2. Añadir 50 ml de solución etanólica de hidróxido de potasio 2N, adaptar el refrigerante de reflujo y calentar en baño María con ligera ebullición, agitando enérgica e ininterrumpidamente hasta que se produzca la saponificación (la solución se vuelve límpida). Calentar, durante 20 minutos más y, a continuación, añadir 50 ml de agua destilada por la parte superior del refrigerante; separar el refrigerante y enfriar el matraz a 30° C aproximadamente.
5.1.3. Transvasar cuantitativamente el contenido del matraz a un embudo de separación de 500 ml, mediante varios lavados con un total aproximado de 50 ml de agua destilada. Agregar 80 ml aproximadamente de éter etílico, agitar enérgicamente durante unos 30 segundos y dejar reposar hasta la separación de las fases (nota 1).
Separar la fase acuosa inferior pasándola a un segundo embudo de separación. Efectuar otras dos extracciones de la fase acuosa por el mismo procedimiento, utilizando cada vez de 60 a 70 ml de éter etílico.
Nota 1: Las posibles emulsiones podrán eliminarse añadiendo pequeñas cantidades de alcohol etílico o metílico con un pulverizador.
5.1.4. Reunir las fracciones etéreas en un mismo embudo de separación y lavarlas con agua destilada (50 ml cada vez) hasta que el agua de lavado presente reacción neutra.
Una vez eliminada el agua de lavado, secar con sulfato sódico anhidro y filtrar sobre sulfato sódico anhidro a un matraz de 250 ml previamente pesado, lavando el embudo y el filtro con pequeñas cantidades de éter etílico.
5.1.5. Destilar el éter hasta que queden unos pocos ml; a continuación, secar con un vacío ligero o en una corriente de nitrógeno, completando el secado en una estufa a 100 °C durante 15 minutos aproximadamente; dejar enfriar en un desecador y pesar.
5.2. Separación de la fracción de esteroles.
5.2.1. Preparación de las placas básicas: sumergir completamente las placas con sílica gel (4.4) en la solución etanólica 0,2N de hidróxido de potasio (4.5) durante 10 segundos; dejar secar las placas en campana durante dos horas y, por último, mantenerlas en una estufa regulada a 100° C durante una hora. Sacarlas de la estufa y conservarlas en un desecador de cloruro de calcio hasta el momento del uso (las placas sometidas a este tratamiento deberán utilizarse en un plazo de quince días como máximo).
Nota 2: Si se utilizan placas básicas de sílica gel para la separación de la fracción de esteroles, ya no es necesario tratar el insaponificable con alúmina. De este modo, todos los compuestos de naturaleza ácida (ácidos grasos y otros) quedan retenidos en la línea de aplicación, y la banda de los esteroles aparece perfectamente diferenciada de la banda de los alcoholes alifáticos y triterpénicos.
5.2.2. Introducir en la cubeta de desarrollo de las placas una mezcla de tolueno-acetona 95:5 (v/v) hasta una altura de 1 cm aproximadamente. Puede utilizarse como alternativa una mezcla de hexano y éter etílico 65:35 (v/v). Cerrar la cubeta con su correspondiente tapa y dejar transcurrir media hora como mínimo, de forma que se alcance el equilibrio líquido-vapor. En las caras interiores de la cubeta pueden colocarse tiras de papel de filtro que se sumerjan en el eluyente: de esta manera el tiempo de desarrollo se reduce casi un tercio y se obtiene una elución más uniforme y regular de los componentes.
Nota 3: Para que las condiciones de elución sean perfectamente reproducibles, la mezcla de desarrollo deberá cambiarse en cada prueba.
5.2.3. Preparar una solución de insaponificable (5.1.5) en cloroformo al 5 % aproximadamente y, con la microjeringa de 100 ml, depositar 0,3 ml de dicha solución en una placa cromatográfica (5.2.1) a unos 2 cm de uno de los bordes, formando una línea lo más fina y uniforme posible. A la altura de la línea de aplicación se depositan, en un extremo de la placa, de 2 a 3 ml de la solución de referencia de esteroles (4.11) para poder identificar la banda de esteroles una vez efectuado el desarrollo.
5.2.4. Introducir la placa en la cubeta de desarrollo, preparada como se indica en el punto 5.2.2. Deberá mantenerse una temperatura ambiente entre 15 y 20°C. Tapar inmediatamente la cubeta y dejar que se produzca la elución hasta que el frente del disolvente se sitúe a 1 cm aproximadamente del borde superior de la placa. Sacar la placa de la cubeta y evaporar el disolvente en una corriente de aire caliente o dejando la placa bajo una campana unos minutos.
5.2.5. Pulverizar la placa ligera y uniformemente con la solución de 2.7diclorofluoresceína. Al examinar la placa a la luz ultravioleta puede identificarse la banda de los esteroles mediante comparación con la mancha obtenida a partir de la solución de referencia; marcar con lápiz negro los límites de la banda a lo largo de los márgenes de fluorescencia.
5.2.6. Rascar con una espátula metálica el gel de sílice contenido en el área delimitada. Introducir el material obtenido, finamente triturado, en el embudo filtrante (3.7); añadir 10 ml de cloroformo caliente, mezclar cuidadosamente con la espátula metálica y filtrar en vacío, recogiendo el filtrado en el matraz cónico (3.8) acoplado al embudo filtrante
Lavar el residuo en el embudo tres veces con éter etílico (empleando cada vez unos 10 ml), recogiendo el filtrado en el mismo matraz cónico acoplado al embudo. Evaporar el filtrado hasta obtener un volumen de 4 a 5 ml, transvasar la solución residual al tubo de ensayo de 10 ml (3.9) previamente pesado, evaporar hasta sequedad mediante calentamiento suave en corriente ligera de nitrógeno, recoger con algunas gotas de acetona, evaporar de nuevo hasta sequedad, introducir en una estufa a 105 °C durante unos 10 minutos, dejar enfriar en el desecador y pesar.
El residuo que queda en el tubo de ensayo está formado por la fracción de esteroles.
5.3. Preparación de los trimetilsililéteres.
5.3.1. Agregar al tubo que contiene la fracción de esteroles el reactivo de silanización formado por una mezcla de piridina-hexametildisilazano-trimetilclorosilano 9:3:1 (v/v/v) (nota 4), a razón de 50 ml por miligramo de esteroles, evitando toda absorción de humedad (nota 5).
Nota 4: Existen soluciones comerciales listas para el uso. Además, también existen otros reactivos de silanización, como el bistrimetilsililtrifluoroacetamida + 1 % de trimetilclorosilano, que se diluye en el mismo volumen de piridina anhidra.
5.3.2. Tapar el tubo y agitar cuidadosamente (sin invertir) hasta la completa disolución de los esteroles. Dejar reposar un cuarto de hora, como mínimo, a temperatura ambiente y centrifugar durante algunos minutos; la solución límpida queda lista para el análisis mediante cromatografía de gases.
Nota 5: La formación de una ligera opalescencia es normal y no ocasiona ninguna interferencia. La formación de una floculación blanca o la aparición de una coloración rosa son indicios de presencia de humedad o de deterioro del reactivo. En este caso deberá repetirse la prueba.
5.4. Cromatografía de gases.
5.4.1. Operaciones preliminares: acondicionamiento de la columna.
5.4.1.1. Colocar la columna en el cromatógrafo uniendo uno de los extremos de la columna al inyector y el otro al detector.
Efectuar los controles generales del equipo para cromatografía de gases (estancamiento de los circuitos de gases, eficacia del detector, eficacia del sistema de división de flujo y del sistema de registro, etc.).
5.4.1.2. Si la columna se utiliza por primera vez, es conveniente acondicionarla previamente. Hacer pasar un ligero flujo de gas a través de la columna, encender el equipo de cromatografía de gases e iniciar un calentamiento gradual hasta alcanzar una temperatura al menos 20 °C superior a la temperatura de trabajo (nota 6). Mantener dicha temperatura durante 2 horas como mínimo; a continuación, poner el equipo completo en condiciones de funcionamiento (regulación del flujo de gases y de la relación de "split", ignición de la llama, conexión con el registrador electrónico, regulación de la temperatura del horno del detector y del inyector, etc.) y registrar la señal con una sensibilidad al menos dos veces superior a la prevista para el análisis. El trazado de la línea de base debe ser lineal, exento de picos de cualquier tipo y no debe presentar deriva.
Una deriva rectilínea negativa indica que las conexiones de la columna no son totalmente estancas; una deriva positiva indica que el acondicionamiento de la columna es insuficiente.
Nota 6: La temperatura de acondicionamiento deberá ser siempre, como mínimo 20 °C inferior a la temperatura máxima prevista para la fase estacionaria utilizada.
5.4.2. Elección de las condiciones de trabajo.
5.4.2.1. Las condiciones de trabajo exigidas son las siguientes:
- temperatura de la columna: 260 °C +/- 5 °C.
- temperatura del evaporador: 280 °C
- temperatura del detector: 290 °C.
- velocidad lineal del gas portador: helio 20 a 35 cm/s, hidrógeno 30 a 50 cm/s.
- relación de "split" de 1/50 a 1/100.
- sensibilidad del instrumento: de 4 a 16 veces la atenuación mínima.
- sensibilidad de registro: 1 a 2 mV f.e.
- velocidad del papel: 30 a 60 cm/hora.
- cantidad de sustancia inyectada: 0,5 a 1 ml de solución de TMSE.
Estas condiciones pueden modificarse en función de las características de la columna y del cromatógrafo, de modo que se obtengan cromatogramas que cumplan los siguientes requisitos:
- el tiempo de retención del b-sitosterol debe ser de 20 +/- 5 minutos.
- el pico del campesterol debe ser: para el aceite de oliva (contenido medio del 3 %), 15 +/- 5 % del total de la escala; para el aceite de soja (contenido medio del 20 %), 80 +/- 10 % del total de la escala.
- se deben separar todos los esteroles presentes; es necesario que los picos no sólo se separen sino que se resuelvan completamente, es decir, que el trazo del pico llegue a la línea de base antes de que se inicie el pico siguiente. No obstante, podrá admitirse una resolución incompleta si el pico a TRR 1,02 puede cuantificarse utilizando la perpendicular.
5.4.3. Realización del análisis.
5.4.3.1. Con la microjeringa de 10 ml tomar 1 mI de hexano, aspirar 0,5 ml de aire y, a continuación, entre 0,5 y 1 mI de la solución problema; elevar el émbolo de la jeringa de modo que la aguja quede vacía. Introducir la aguja a través del septum y, después de 1 o 2 segundos, inyectar rápidamente; transcurridos unos 5 segundos, extraer la aguja lentamente.
5.4.3.2. Continuar el registro hasta la completa elución de los TMSE de los esteroles presentes.
La línea de base debe ajustarse en todo momento a las condiciones exigidas (5.4.1.2).
5.4.4. Identificación de los picos.
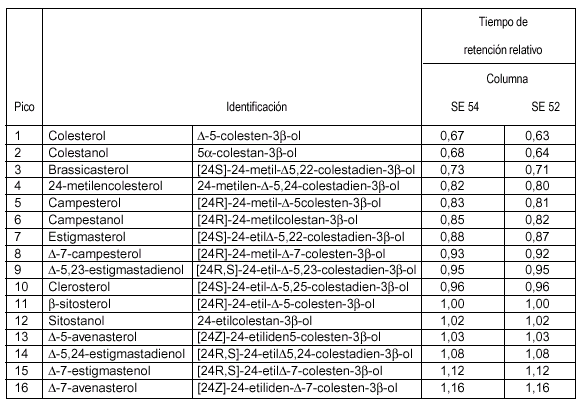
Para la identificación de los diferentes picos se utilizan los tiempos de retención y la comparación con mezclas de TMSE de los esteroles analizadas en las mismas condiciones. La elución de los esteroles se efectúa en el orden siguiente: colesterol, brassicasterol, 24metilencolesterol, campesterol, campestanol, estigmasterol, D-7-campesterol, D-5,23estigmastadienol, clerosterol, b-sitosterol, sitostanol, D-5-avenasterol, D-5,24estigmastadienol, D-7-estigmastenol, D-7-avenasterol.
En el cuadro I figuran los tiempos de retención correspondientes al b-sitosterol para las columnas SE 52 y SE 54.
5.4.5. Determinación cuantitativa.
5.4.5.1. Calcular las áreas de los picos del a-colestanol y de los esteroles utilizando el integrador. No se tomarán en cuenta los picos de aquellos componentes que no figuren en el cuadro 1. El factor de respuesta para el a-colestanol debe considerarse como 1.
5.4.5.2. Calcular del modo siguiente el contenido de cada uno de los esteroles, expresado en mg/ 100 g de materia grasa:
|
esterol x = |
Ax . ms . 100 |
|
. |
As . m |
siendo:
Ax: área del pico del esterol x, en milímetros cuadrados.
As: área del pico del a-colestanol.
ms: peso de a-colestanol añadido, en miligramos.
m: peso de la muestra tomado para la determinación, en gramos.
6. EXPRESION DE LOS RESULTADOS
6.1. Se registra el contenido de cada uno de los esteroles en mg/100 g de materia grasa y, su suma como "esteroles totales".
6.2. El porcentaje de cada uno de los esteroles simples es la razón entre el área del pico correspondiente y la suma de las áreas de los picos de los esteroles.
|
% del esterol x = |
Ax . |
100 |
|
. |
SA |
. |
siendo: Ax: área del pico de x.
SA: suma de las áreas de todos los picos.
APENDICE
Determinación de la velocidad lineal del gas.
Inyectar en el cromatógrafo, preparado para trabajar en condiciones normales, de 1 a 3 ml de metano (o propano) y medir el tiempo que tarda el gas en recorrer la columna, desde el momento de la inyección hasta el momento en que aparece el pico (t M ). La velocidad lineal en cm/s viene dada por L/ t M , siendo L la longitud de la columna en cm y t M el tiempo, expresado en segundos.
CUADRO I
Tiempos de retención relativos de los esteroles
Art. 4° — Inclúyese en el artículo 1413 del Código Alimentario Argentino, el apartado 11.23, el cual quedará redactado de la siguiente forma:
"11.23 DETERMINACION DEL CONTENIDO DE ACIDOS GRASOS TRANS MEDIANTE CROMATOGRAFIA GASEOSA CON COLUMNA CAPILAR
1. OBJETIVO Y CAMPO DE APLICACION
El presente método describe un procedimiento para la determinación del contenido de ácidos grasos trans en las materias grasas vegetales mediante cromatografía gaseosa con columna capilar. El método es aplicable a aceites y grasas que contienen ácidos grasos entre 10 y 24 átomos de carbono. Esto no es aplicable a grasas vegetales que hayan experimentado algún proceso de hidrogenación.
2. PRINCIPIO
La técnica consiste en la preparación de ésteres metílicos de los ácidos grasos provenientes de la materia grasa; el análisis mediante cromatografía gaseosa; la identificación de los ácidos grasos por sus tiempos de retención y la cuantificación mediante la determinación de la relación del área bajo el pico correspondiente respecto de la suma de las áreas de los picos del total de ácidos grasos.
Las condiciones de trabajo y materiales detallados a continuación podrán adecuarse a las características de la columna, el cromatógrafo y la disponibilidad de insumos con que cuente el laboratorio.
3. FUNDAMENTO
Determinación de los ácidos grasos trans insaturados de 18 átomos de carbono que pueden estar presentes en concentraciones específicas en aceites y grasas vegetales naturales, como en aceites que hayan experimentado un proceso de refinación.
4. DEFINICIONES
Dependiendo del método, el contenido de ácidos grasos trans de los aceites o grasas vegetales, es obtenido mediante la suma del contenido relativo de los siguientes ácidos grasos respecto del total de ácidos grasos:
- trans-octadecenoico (T 18:1)
- trans, trans-octadecadienoico (TT 18:2)
- cis-tans y trans-cis octadecadienoico [(CT + TC) 18:2]
- trans-cis-trans, cis-cis-trans, cis-trans-cis y trans-cis-cis octadecatrienoico [(TCT + CCT + CTC + TCC) 18:31]
5. MATERIALES
5.1. Cromatógrafo gaseoso que pueda funcionar con columna capilar, provisto de un sistema de división de flujo formado por:
5.1.1. Horno para la columna, que pueda mantener la temperatura deseada con precisión de +/- 1 °C.
5.1.2. Unidad de inyección con temperatura controlada.
5.1.3. Detector de ionización de llama y convertidor-amplificador.
5.1.4. Contador de flujo para el gas portador y los gases auxiliares.
5.2. Columna capilar de vidrio o sílice fundida, de 50 m de longitud y de 0,25 a 0,32 mm de diámetro interno, recubierta con cianopropilsilicona o equivalente, con un espesor uniforme que oscile entre 0,10 y 0,30 mm.
5.3. Registrador-integrador que pueda funcionar con el convertidor-amplificador, con un tiempo de respuesta no superior a 1 segundo y con velocidad de papel variable.
5.4. Microjeringa de 10 ml para cromatografía de gases, con aguja endurecida.
5.5. Evaporador rotatorio.
5.6. Material necesario para la preparación de los ésteres metílicos.
6. REACTIVOS
6.1. Gas portador: helio o hidrógeno para cromatografía de gases.
6.2. Gases auxiliares: aire e hidrógeno, para cromatografía de gases.
6.3. Muestras de referencia: ésteres metílicos de ácidos grasos puros, en especial los isómeros cis y trans de los ácidos octadecenoico, octodecadienoico y octadecatrienoico 6.4. n-hexano.
7. PROCEDIMIENTO
7.1. Preparación de los ésteres metílicos. Usar un procedimiento que incluya una catálisis básica. Las grasas y aceites con un grado de acidez libre mayor al 3% deben ser neutralizados antes de su utilización.
7.2. Control del cromatógrafo gaseoso y el acondicionamiento de la columna.
Realizar los controles preliminares de la unidad de inyección y el registrador. Ajustar la columna en el cromatógrafo gaseoso sin conectar al detector. Hacer pasar un flujo ligero de gas portador (1 a 2 ml/minuto) a través de la columna y calentar gradualmente hasta los 210 °C en no menos de 4 horas. Mantener la columna a esta temperatura por al menos una noche y luego dejar enfriar. Conectar el detector, controlar la estabilidad de la conexión y llevar la unidad de inyección a la temperatura de acondicionamiento. Encender el detector y el registrador, luego de 3 o 4 horas, controlar que la linealidad y la deriva de la línea de base sean satisfactorias, registrar la señal con una sensibilidad por lo menos cuatro veces mayor que la prevista para el análisis (una deriva positiva mayor a 5%/hora del total de la escala indica que la columna no fue correctamente acondicionada).
7.3. Cambio de las condiciones de operación y control de la eficacia de la columna.
7.3.1. Condiciones de operación.
Las condiciones generales son las siguientes:
- temperatura de la columna: 150 - 200 °C, de ser posible, programar el equipo para obtener un aumento de la temperatura de 5°C/minuto.
- temperatura del inyector: 250 °C.
- temperatura del detector: 260 - 280 °C.
- volumen del gas portador (helio o hidrógeno): 1,2 ml/minuto
- cantidad de sustancia inyectada: 1 ml de solución al 2% en hexano.
Estas condiciones pueden modificarse en función de las características de la columna y del cromatógrafo. Controlar que el pico correspondiente al ácido linolénico (C18:3) aparezca como un pico definido, justo por delante del pico del ácido eicosanoico (C20:1). Si esto no ocurre, puede realizarse el cambio de la temperatura del horno y/o usar columnas de diferente polaridad.
7.3.2. Eficiencia de la columna.
7.3.2.1. La eficiencia de la columna se determina mediante su poder de separación y resolución. Por lo tanto, si los análisis fueron realizados correctamente, deben obtenerse cromatogramas que alcancen los siguientes requerimientos:
- poder de separación: los picos correspondientes a los ésteres metílicos de todos los ácidos grasos presentes en la muestra deben ser absolutamente distinguibles.
- poder de resolución: los picos deben estar completamente resueltos. Por ejemplo, el trazado del pico debe retomar a la línea de base antes del comienzo del trazado del pico siguiente. Para algunos pares de picos típicos, es tolerable una resolución incompleta, por ejemplo, los ácidos cis C18:1 y trans C18:1.
En algunos casos, la columna es considerada satisfactoria si el índice de resolución es mayor a 2. El índice de resolución entre dos picos se define por la razón:
a/b
Donde:
a: distancia desde la línea de base hasta la cima del pico más bajo.
b: distancia desde la línea de base hasta el punto más bajo del trazado entre dos picos.
7.3.2.2. Para controlar que los requerimientos arriba mencionados sean satisfactorios, inyectar varias veces cantidades iguales de la porción de ésteres metílicos a analizar ajustando las condiciones de temperatura y flujo del gas portador, hasta obtener la mejor separación entre los picos. Para controlar el poder de resolución, reducir la porción a analizar, y aumentar la sensibilidad hasta obtener la mejor resolución del pico.
7.4. Procedimiento analítico.
Con la microjeringa de 10 ml tomar 1 ml de hexano, aspirar 0,5 mI de aire y, a continuación, entre 0,5 y 1 mI de la solución problema; elevar el émbolo de la jeringa de modo que la aguja quede vacía. Introducir la aguja a través del septum y, después de 1 o 2 segundos, inyectar rápidamente; transcurridos unos 5 segundos, extraer la aguja lentamente. Continuar el registro hasta la completa elución de los ésteres metílicos. La línea de base debe ajustarse en todo momento a las condiciones exigidas (7.3.2.l.).
7.5. Identificación de los picos.
El orden de aparición de los ésteres metílicos en el cromatograma es función directa del número de átomos de carbono. Los ésteres insaturados eluyen a continuación del éster saturado correspondiente y su elución se relaciona con la cantidad de dobles enlaces que contienen. Los ésteres de los ácidos grasos trans eluyen antes del isómero cis correspondiente.
Cada uno de los ésteres metílicos se identifican usando su tiempo de retención el cual se compara con los tiempos de retención de la referencia.
7.6. Determinación cuantitativa.
7.6.1. Calcular las áreas de cada pico utilizando el integrador. Para asegurar la correcta sensibilidad analítica, la altura del pico correspondiente al éster metílico del ácido araquídico no debe ser menor al 20% del total de la escala. En el caso de aceite de maní, la altura del pico no debe ser menor al 50%.
7.6.2. El porcentaje de cada ácido graso se calcula como la razón entre el área bajo el pico correspondiente al ácido graso y la suma de las áreas bajo todos los picos usando la siguiente fórmula:
|
% ácido graso x = |
Ax .100 |
|
. |
SA |
siendo:
Ax: área del pico del ácido graso x
SA: suma de las áreas de todos los picos
8. EXPRESION DE LOS RESULTADOS
8.1. Registrar el % total de ácidos grasos trans, como la suma de los contenidos porcentuales de los siguientes ácidos grasos:
- trans-octadecenoico
- trans-trans octadecadienoico
- cis-trans y trans-cis octadecadienoico
- trans-cis-trans, cis-cis-trans, cis-trans-cis y trans-cis-cis octadecatrienoico
Expresar los resultados con dos cifras decimales.
NOTA
Se requiere tomar ciertas precauciones para asegurar una apropiada evaluación analítica de los resultados. Cuando se introduce la muestra dentro del inyector del cromatógrafo se forman pequeñas cantidades de ácido trans oleico. Para prevenir esto, es importante realizar el control de la siguiente manera:
Preparar una muestra de ésteres metílicos de aceite de oliva virgen extra de origen conocido (o un estándar de oleato de metilo, o una mezcla con ésteres metílicos de ácidos grasos que contengan una mínima cantidad de ácido oleico y una cantidad certificada de ácidos grasos trans del 60%) como se describe en la sección 7.1. y analizar bajo las condiciones especificadas en el método. Se mide el porcentaje de ácidos grasos trans octadecenoico. Sin embargo, se tolera una presencia de ácidos grasos octadecenoico del 0,01%.
Si se registran valores mayores, debería reemplazarse el disco del inyector por otro perfectamente limpio, y el análisis debería repetirse.
Deben tomarse mayores precauciones en el caso que el disco del inyector esté empaquetado con una variedad de materiales (lana de vidrio, tierra de diatomea, etc.) puesto que la presencia de tales materiales puede catalizar la formación de los ácidos grasos trans.
9. PRECISION
La precisión del método se evalúa sobre la base de ensayos inter-laboratorios, involucra la participación de 10 laboratorios que realizan el análisis de 3 muestras, una de aceite de oliva con 20% de aceite de soja, otra de aceite de oliva virgen con un 10% de aceite de soja y otra de aceite de soja; por duplicado. Con los resultados obtenidos se construye una tabla de parámetros de precisión.
9.1. Repetibilidad.
Cuando el método se aplica correctamente, la diferencia entre dos resultados obtenidos del mismo material por distintos operadores, usando el mismo equipamiento dentro de intervalos cortos de tiempo, no debería ser mayor al promedio del límite de repetibilidad calculado en el ensayo interlaboratorios, con una probabilidad del 95%.
9.2. Reproducibilidad.
Cuando se aplica el método de la manera correcta, la diferencia entre los resultados de dos ensayos simples obtenidos con materiales idénticos en laboratorios diferentes, con diferentes operadores y equipos, no debería ser mayor en promedio que los límites de reproducibilidad dados del ensayo inter-laboratorios con una probabilidad del 95%".
Art. 5° — La presente Resolución comenzará a regir a partir de los 60 (SESENTA) días de su publicación en el Boletín Oficial.
Art. 6° — Regístrese, comuníquese, dése a la Dirección Nacional del Registro Oficial para su publicación. Cumplido, archívese PERMANENTE. — Carlos E. Filgueira Lima. — Haroldo A. Lebed.