100. CROMATOGRAFIA
La cromatografía es un método por el cual las sustancias se separan mediante un proceso de migración diferencial en un sistema que consta de dos fases. Una fase que fluye continuamente en una dirección dada (fase móvil) y otra que permanece fija (fase estacionaria). En estos sistemas los componentes de una mezcla pueden presentar diferentes movilidades debido a diferencias en la capacidad de adsorción, partición, solubilidad, presión de vapor, tamaño molecular o carga. Los mecanismos de separación son: adsorción, disolución y partición, filtración y permeación o tamices moleculares, intercambio iónico.
Las técnicas aplicadas mediante los distintos mecanismos-mencionados empleados en esta Farmacopea son: Cromatografía en columna, Cromatografía en papel, Cromatografía en capa delgada, Cromatografía de gases, Cromatografía líquida de alta eficacia y Cromatografía de exclusión.
La Cromatografía de adsorción se basa en la separación de un soluto entre la fase estacionara constituida por un adsorbente, como por ej., alúmina activada, sílica gel y resinas de intercambio iónico y la fase móvil constituida por el solvente de elución.
La Cromatografía de partición se basa en la distribución selectiva del soluto entre la fase estacionaria y la fase móvil. Se clasifica en cromatografía de partición en fase normal y cromatografía de partición en fase reversa. En la cromatografía de partición en fase normal las sustancias a separar se distribuyen entre dos líquidos inmiscibles uno de los cuales es más polar, actúa como fase estacionaria y se encuentra adsorbido sobre un soporte sólido, brindando una gran superficie de contacto a la fase móvil menos polar. En la cromatografía de partición en fase reversa la fase estacionaria es menos polar que la fase móvil.
El grado de partición de un compuesto dado entre las dos fases líquidas se expresa por su coeficiente de partición o de distribución y puede modificarse variando la composición de la fase móvil. En el caso de compuestos que se disocian, la distribución se puede controlar modificando, entre otras propiedades, el pH, la constante dieléctrica y la fuerza iónica.
La Cromatografía de intercambio iónico se emplea para separar compuestos ionizables y solubles en agua. Las fases estacionarias empleadas son generalmente resinas orgánicas sintéticas. Las resinas de intercambio catiónico contienen sitios activos con carga negativa y se emplean para separar compuestos básicos, como por ej., las aminas. Las resinas de intercambio aniónico tienen sitios activos con carga positiva para la separación de compuestos ácidos, corno por ej., fosfatos, sulfonatos o carboxilatos. Los compuestos iónicos o ionizables, solubles en agua, son atraídos a las resinas y las diferencias en la afinidad producen la separación cromatográfica. El pH de la fase móvil, la temperatura, el tipo de ion, la concentración iónica y los modificadores orgánicos afectan el equilibrio; estas variables pueden ajustarse para obtener el grado de separación deseado.
La Cromatografía por tamices moleculares se basa en el intercambio repetido de los compuestos con la fase móvil y con la fase líquida estacionaria que se encuentra dentro de los poros del material de relleno.
Empleo de Sustancias de referencia en ensayos de identificación - En Cromatografía en papel y en Cromatografía en capa delgada, la relación entre la distancia recorrida por una sustancia y la distancia recorrida por el frente de la fase móvil, se denomina relación de frente, Rf, de la sustancia. La relación entre la distancia recorrida por una sustancia y la distancia recorrida por una Sustancia de referencia, se denomina RE de la sustancia.
En el caso de la Cromatografía en papel se han observado diferencias en el valor de Rf cuando los cromatogramas se desarrollan en dirección paralela a las fibras de papel en comparación con los desarrollados en forma perpendicular a dicha dirección. En consecuencia, la orientación de las fibras del papel en lo que se refiere al flujo de la fase móvil debe ser la misma para una serie de cromatogramas. [NOTA: por lo general, el fabricante indica el sentido de las fibras en los envases de papel para Cromatografía].
Los valores absolutos de Rf, son difíciles de establecer, ya que varían con las condiciones experimentales por lo tanto se logra una mejor identificación cuando se emplea una muestra de la sustancia a ensayar como Sustancia de referencia. Para este fin se preparan soluciones de la muestra, la Sustancia de referencia y una mezcla de partes iguales de ambas y se aplican sobre una línea paralela a uno de los bordes de la placa cromatográfica u hoja de papel. Cada aplicación contiene aproximadamente la misma cantidad, en peso, de la muestra y la Sustancia de referencia. Si la misma y la Sustancia de referencia son idénticas, todos los cromatogramas deben coincidir en color y valor de Rf, y el cromatograma de la mezcla debe mostrar una única mancha.
En Cromatografía en columna, Cromatografía en papel y Cromatografía en capa delgada, las sustancias pueden ser localizadas por: (a) observación directa con luz visible o luz ultravioleta, si las sustancias poseen color o producen fluorescencia; (b) observación con luz visible o ultravioleta, después de agregar un reactivo que reaccione con las sustancias separadas; (c) mediante un contador Geiger-Müller o con técnica autorradiográfica, cuando se trabaja con sustancias radiactivas o (d) por estimulación o inhibición del desarrollo microbiano, colocando trozos de papel que contienen las sustancias separadas en medios de cultivo apropiados.
CROMATOGRAFIA EN COLUMNA
La Cromatografía en columna se emplea para la separación de sustancias en escala preparativa.
Cromatografía de adsorción
Preparación de la columna - Emplear un tubo cromatográfico, cilíndrico, de vidrio o del material especificado en la monografía correspondiente, generalmente de 10 a 30 mm de diámetro interno y de 150 a 400 mm de largo. En su extremo inferior, el tubo se angosta formando un tubo de salida que generalmente posee un diámetro interno de 3 a 6 mm, pudiendo incluir un robinete para el control exacto del caudal. Generalmente se emplea una varilla de vidrio u otro material para colocar un trozo de lana de vidrio o algodón, en la base del tubo, y si fuera necesario, compactar el adsorbente o una suspensión del mismo uniformemente dentro del tubo. En algunos casos, en la base del tubo se encuentra soldado un disco de vidrio poroso que actúa como soporte del contenido del tubo. Colocar el adsorbente especificado en la monografía correspondiente de manera que se forme una columna compacta, homogénea y sin fisuras.
Los adsorbentes más empleados son alúmina, gel de sílice activado y tierra de diatomeas.
Procedimiento - Disolver la muestra en una cantidad apropiada de solvente y agregarla por el extremo superior de la columna. Dejar que esta solución se adsorba y luego agregar nuevas porciones de solvente, de manera que fluya a través de la columna espontáneamente, por aplicación de vacío en la base o ejerciendo presión en el extremo superior. En algunos casos, puede modificarse el procedimiento de carga de la muestra en la columna. Si el producto es sólido (como por ej., comprimidos pulverizados) se lo mezcla íntimamente con una porción del adsorbente empleado para rellenar la columna, sin necesidad de separarlo de su excipiente y se agrega esta mezcla al extremo superior de la columna. El paso posterior de solvente hace progresar la sustancia a través de la columna.
La separación y aislamiento puede mejorarse haciendo circular mayores cantidades de fase móvil o un solvente de mayor poder eluyente, a través de la columna y recolectando distintas fracciones del eluato que contienen los componentes de la muestra.
La eficiencia de la separación suele controlarse realizando un cromatograma en capa delgada de las fracciones individuales.
Cromatografía de partición
Preparación de la columna - Emplear un tubo cromatográfico de aproximadamente 22 mm de diámetro interno y 200 a 300 mm de largo, sin disco de vidrio poroso en su extremo, al que se ajusta un tubo de salida, sin robinete, de aproximadamente 4 mm de diámetro interno y 50 mm de largo, a menos que se especifique de otro modo en la monografía correspondiente. Adaptar un trozo de lana de vidrio en la base del tubo. Transferir el volumen de fase estacionaria y la cantidad de soporte sólido especificados en la monografía correspondiente a un vaso de precipitados de 100 a 250 ml y mezclar hasta obtener una mezcla homogénea y espesa. Transferir esta mezcla al tubo cromatográfico y apisonar presionando suavemente, hasta obtener una masa uniforme. Si la cantidad de soporte sólido especificada es mayor de 3 g, transferir la mezcla al tubo en porciones de aproximadamente 2 g y apisonar cada porción.
Procedimiento - La muestra se puede agregar a la parte superior de la columna disuelta en un volumen apropiado de fase móvil o empleando una solución de la muestra en un volumen apropiado de fase estacionaria mezclada con una parte adicional del soporte sólido y transferida a la parte superior de la columna como una capa extra de soporte. La muestra puede también ser incorporada en la fase estacionaria, completando la transferencia cuantitativa al tubo cromatográfico, lavando el vaso de precipitados, empleado para la preparación de la muestra, y agregando una mezcla de aproximadamente 1 g de soporte sólido y varías gotas del solvente empleado para preparar la solución muestra. Colocar un trozo de lana de vidrio fina por encima del soporte de la fase estacionaria para completar la columna. Dejar que se adsorba completamente en la fase estacionaria y luego agregar fase móvil en varias porciones, permitiendo que cada una penetre en la columna completamente, antes de comenzar la elución. Como fase móvil emplear el solvente o la solución especificada en la monografía correspondiente. Equilibrar la fase móvil con agua si la fase estacionaria es una solución acuosa o si la fase estacionaria es un líquido orgánico polar, equilibrar con ese líquido.
Cuando la valoración o el ensayo requieren el empleo de varias columnas cromatográficas colocadas en serie, cuando se especifique el agregado de la fase móvil en porciones o el cambio en la composición de la misma, dejar que cada porción drene completamente a través de la columna y lavar el vástago con fase móvil antes del agregado de cada porción.
CROMATOGRAFIA EN PAPEL
El mecanismo predominante en Cromatografía en papel es la partición, ésto se debe a que el papel posee un contenido natural de agua que puede ser considerada como fase estacionaria. Sin embargo, en la práctica, las separaciones frecuentemente son el resultado de la combinación de efectos de adsorción y partición.
Cromatografía descendente
Aparato - Consta de:
- Una cámara con cierre hermético para permitir la saturación con los vapores de la fase móvil, generalmente construida de vidrio, acero inoxidable o porcelana y diseñada de manera que permita seguir el proceso sin necesidad de abrirla.
- Un bastidor de material resistente a la corrosión, aproximadamente 5 cm más corto que la altura interior de la cámara. El bastidor sirve de soporte a las cubetas que contienen la fase móvil y de las cuales se suspenden las hojas de papel.
- Una o más cubetas de vidrio o de material inatacable por la fase móvil.
- Varillas de vidrio, colocadas en forma paralela al borde de cada cubeta para mantener suspendidas la hoja de papel.
- Hojas de papel, cromatográfico de textura y espesor apropiados.
Procedimiento - Trazar una línea transversal, cerca de uno de los extremos del papel, de modo que cuando se sumerja en la fase móvil quede a unos pocos centímetros por debajo de la varilla de vidrio. Disolver la muestra en un solvente apropiado. Aplicar un volumen de la solución así obtenida que contenga aproximadamente entre 1 y 20 mg de la sustancia a ensayar sobre la línea anteriormente trazada y un volumen similar de la Sustancia de referencia dejando no menos de 3 cm entre cada aplicación. La aplicaciones no deben formar una mancha mayor de 6 a 10 mm de diámetro, para ello aplicar las soluciones en porciones sucesivas dejando secar luego de cada aplicación.
Sujetar el papel por el extremo donde se aplicó la muestra dentro de la cubeta. El papel debe pasar por encima de la varilla colocada en el borde de la cubeta y debe colgar libremente en la cámara, sin tocar su fondo o sus paredes.
Colocar una cantidad apropiada de fase móvil en el fondo de la cámara y cerrarla herméticamente. Dejar la cámara en estas condiciones durante un período que permita que el ambiente se sature y el papel se equilibre con el vapor de la fase móvil. La saturación de la cámara puede favorecerse mediante el revestimiento de las paredes internas con un papel humedecido en fase móvil.
Colocar la fase móvil en la cubeta y cerrar la cámara herméticamente. Dejar descender la fase móvil por el papel, hasta que haya recorrido la distancia deseada. Retirar el papel de la cámara, marcar rápidamente el frente del solvente y dejar secar.
Observar los cromatogramas directamente o revelar la posición de las manchas empleando reactivos apropiados. Comparar los cromatogramas obtenidos a partir de la muestra y la Sustancia de referencia.
La sección del papel que contiene la sustancia o sustancias aisladas puede cortarse y eluirse con un solvente apropiado. La solución resultante puede ser valorada mediante técnicas químicas o instrumentales apropiadas empleando Sustancias de referencia, tratadas de la misma manera que la muestra, en un intervalo apropiado dé concentraciones para realizar una curva de calibración.
Cromatografía ascendente
Aparato - Emplear el aparato descripto en Cromatografía descendente.
Procedimiento - Aplicar la muestra y la Sustancia de referencia según se indica para el Procedimiento en Cromatografía descendente. Transferir una cantidad apropiada de fase móvil para cubrir el fondo de la cámara. Colocar la cubeta vacía en el fondo de la cámara. Colocar el papel empleando un soporte apropiado dentro de la cubeta vacía, procurando que no toque las paredes de la cámara. Cerrarla herméticamente y dejarla en estas condiciones durante un período que permita que el ambiente se sature y el papel se equilibre con el vapor de la fase móvil. Colocar la fase móvil en la cubeta y cerrar la cámara herméticamente. Dejar que el solvente ascienda por el papel hasta que haya recorrido la distancia deseada. Retirar el papel de la cámara, marcar el frente del solvente y dejar secar.
[NOTA: pueden emplearse cámaras cilíndricas, de vidrio, que no requieren el empleo de cubetas y en las que la fase móvil se coloca directamente sobre el fondo de la cámara. El papel se suspende de la tapa que cierra la cámara. Durante la etapa de equilibrio, el extremo inferior del papel no debe tocar la fase móvil. La cromatografía comienza haciendo descender la hoja de papel de modo que toque la fase móvil].
CROMATOGRAFIA EN CAPA DELGADA
La Cromatografía en capa delgada es comúnmente empleada para la identificación de sustancias. El mecanismo de separación predominante es la adsorción pero dependiendo del adsorbente empleado pueden observarse también fenómenos de partición.
En cromatografía en capa delgada el adsorbente está constituido por una capa uniforme y relativamente delgada de un material finamente pulverizado que se aplica sobre una placa rígida de vidrio, plástico o metal. Esta técnica presenta varias ventajas sobre la cromatografía en papel, se pueden emplear mayores cantidades de muestra; el tiempo requerido es menor por lo tanto los riesgos de alteración de la muestra por oxidación o por acción de los solventes disminuyen y permite el uso de adsorbentes minerales que hacen posible el empleo de reveladores agresivos, como por ej., ácido sulfúrico.
Aparato - Consta de:
- Placas de material inerte las más empleadas son de vidrio de 20 cm X 20 cm.
- Un bastidor o soporte de material resistente a la corrosión y aproximadamente 5 cm más corto que la altura interna de la cámara destinado a sostener una o más placas que se disponen enfrentadas por su cara no cubierta por el adsorbente.
- Materiales adsorbentes finamente divididos que pueden ser de origen mineral (gel de sílice, alúmina, etc.) u orgánico (celulosa, poliamidas, etc.), normalmente de 5 a 40 mm de diámetro. Pueden aplicarse directamente sobre la placa de vidrio o adherirse a la placa a través de emplasto de parís (sulfato de calcio hidratado) en una relación de 5 a 15% o con pasta de almidón u otros aglutinantes. El primero no produce superficies duras como el almidón, pero no es afectado por reactivos reveladores fuertemente oxidantes. El adsorbente puede contener materiales que ayuden a la visualización de las manchas que absorben la luz ultravioleta.
- Un aparato apropiado para esparcir el adsorbente de modo que al desplazarlo sobre la placa permita aplicar en toda su superficie una capa uniforme con el espesor deseado.
- Una cámara de vidrio cilíndrica o rectangular de aproximadamente 30 cm de altura por 30 cm de ancho y 16 cm de fondo, provista de una tapa del mismo material que permita el cierre hermético para saturarla con los vapores de la fase móvil.
- Fase móvil constituida por mezclas de solventes orgánicos o soluciones acuosas según se indique en la monografía correspondiente.
- Reactivos reveladores específicos indicados en las monografías correspondientes.
- Un pulverizador que permita aplicar el reactivo revelador, resistente al ataque del mismo.
[NOTA: pueden emplearse placas comerciales preparadas o bien prepararlas en el laboratorio].
Preparación de la placa - Limpiar perfectamente las placas por inmersión en mezcla sulfocrómica (ver 1090. Limpieza de materiales de vidrio) y luego lavarlas con abundante agua hasta que el líquido que escurre no deje en la superficie de las placas manchas visibles. Secarlas perfectamente.
Suspender el adsorbente, con el agregado o no de reactivos, como por ej., soluciones reguladoras, sustancias fluorescentes, etc., en agua o en solventes orgánicos volátiles, agitar durante 30 segundos, hasta formar una suspensión homogénea. Extender la suspensión sobre una o varias placas, manualmente o con ayuda del aplicador, hasta lograr una capa uniforme de 0,25 a 1 mm de espesor. Dejar en reposo durante 5 minutos. Secar a 105 °C durante 30 minutos y dejar enfriar en un desecador. Generalmente 30 g de adsorbente y 60 ml de agua son suficientes para cinco placas de 20 cm X 20 cm. Cuando se emplea emplasto de parís como aglutinante completar la aplicación de los adsorbentes dentro de los 2 minutos de haber agregado el agua, ya que posteriormente la mezcla empieza a endurecer.
Cuando las placas estén secas y a temperatura ambiente, verificar la uniformidad de la distribución y la textura de la capa adsorbente; la luz transmitida muestra uniformidad en la distribución y la luz reflejada muestra uniformidad en la textura. Conservar las placas cromatográficas en un desecador. Deben emplearse dentro de los tres días posteriores a su preparación.
Procedimiento - Colocar en la cámara una cantidad suficiente de fase móvil hasta obtener una capa de 1,5 cm. Adherir hojas de papel de filtro embebidas en la fase móvil a las paredes de la cámara, para facilitar la saturación de la misma con el vapor del líquido, a menos que se especifique de otro modo, en la monografía correspondiente. Cerrar la cámara herméticamente. Trazar una línea a 2,5 cm del borde inferior y lateral de la placa. Aplicar por separado sobre la línea trazada anteriormente y a no menos de 2 cm entre cada aplicación la solución muestra y la solución estándar empleando una micropipeta para obtener una mancha lo más pequeña posible (preferentemente con un diámetro mayor de 5 mm o bien en una banda perpendicular al sentido del desarrollo del cromatograma, de 10 a 20 mm de largo y 2 a 6 mm de ancho). La aplicación puede realizarse en porciones sucesivas que permitan acumular la cantidad de material requerido, dejando secar cada vez, antes de efectuar la siguiente aplicación.
Dejar secar las aplicaciones, ubicar la placa en el soporte y colocarla dentro de la cámara, de modo que la fase móvil llegue al borde inferior de la placa. Cerrar la cámara 5, desarrollar, los cromatogramas hasta que el frente del solvente haya recorrido aproximadamente tres cuartos de la longitud de la placa, o la distancia indicada en la monografía correspondiente. Los cromatogramas requieren para su desarrollo aproximadamente de 15 minutos a 1 hora. Retirar la placa de la cámara, marcar el frente del solvente y dejar secar.
En el caso de que se especifique una cromatografía en capa delgada bidimensional, la placa cromatográfica se somete a una cromatografía en una dirección, se seca y luego se somete a una segunda cromatografía en ángulo recto respecto de la dirección original, generalmente en otra cámara equilibrada con un sistema de solventes diferente.
Examinar la placa empleando el método especificado en la monografía correspondiente.
La sección de la placa que contiene la sustancia o sustancias aisladas también pueden separarse empleando una espátula, eluirse con un solvente apropiado y cuantificarse empleando espectrofotometría o fluorescencia.
Existen además instrumentos de lectura, los densitómetros, que miden la concentración de la sustancia sobre la placa como reflectancia o transmitancia, por absorción de luz o fluorescencia, empleando longitudes de onda entre 190 y 800 nm seleccionadas con filtros o sistemas de difracción. La serial generada puede ser enviada a un registrador gráfico, integrador o una computadora provista de programas apropiados.
CROMATOGRAFIA DE GASES
La Cromatografía de gases se emplea para la separación de sustancias o mezcla de sustancias volátiles. Pueden emplearse los siguientes sistemas:
Cromatografía gas-líquido la fase estacionaria puede estar contenida en columnas rellenas o capilares. En las columnas rellenas, la fase líquida se deposita sobre un soporte sólido finamente dividido e inerte en una columna de 1 a 3 m de longitud X 2 a 4 mm de diámetro interno. Los soportes más comúnmente empleados son tierra de diatomeas, polímeros porosos o carbono grafito. En las columnas capilares, que no contienen soporte, la fase líquida se deposita en la superficie interna de la columna o puede unirse químicamente a ella.
Cromatografía gas-sólido se emplea como fase estacionaria alúmina, sílice, carbono o resinas porosas poliaromáticas.
Aparato - Consta de:
- Un reservorio de gas transportador constituido por un gas comprimido, como por ej.: helio, nitrógeno, hidrógeno, argón o mezclas (como por ej., 95% de argón y 5% de metano) según el tipo de detector y columna empleados.
- Un sistema de inyección constituido por una jeringa o un inyector automático.
Los inyectores pueden ser:
Inyectores de flujo dividido: son inyectores capaces de dividir la muestra en dos fracciones, una pequeña que se introduce en la columna y una grande que se desecha. También pueden emplearse en modo normal sin desechar ninguna porción de la muestra para el análisis de trazas o componentes minoritarios.
Inyectores de purga y trampa: están equipados con un dispositivo por el cual las sustancias volátiles de la solución se capturan en una trampa de baja temperatura. Una vez que se completa el atrapado de las sustancias, se liberan en el gas transportador mediante, la calefacción rápida de la trampa, la cual posee un dispositivo programable de temperatura.
Inyectores de espacio libre superior: poseen un sistema de temperatura programable. Las muestras líquidas o sólidas se colocan en envases perfectamente cerrados y se calientan durante un período de tiempo fijo, lo que permite que los componentes volátiles de la muestra alcancen un equilibrio entre las fases no gaseosa y gaseosa (espacio libre superior del envase). Una vez establecido el equilibrio, el inyector introduce automáticamente una cantidad determinada del espacio libre superior del envase en el cromatógrafo de gases.
- Las columnas pueden ser capilares o rellenas. Las columnas capilares, generalmente fabricadas con sílice fundida, poseen un diámetro interno de 0,20 a 0,53 mm (estas últimas también llamadas macrocapilares) y 5 a 60 m de longitud. El espesor de la fase estacionaria, que a veces se une químicamente a la superficie interna, es de 0,1 a 1,0 mm, aunque para las fases etacionarias no polares puede ser hasta 5 mm.
Las columnas rellenas, de vidrio o metal, poseen un diámetro interno de 2 a 4 mm y 1 a 3 m de largo. Generalmente contienen un polímero poroso sobre soporte sólido o solo soporte sólido.
El tiempo de retención y la eficiencia dependen de la temperatura, del caudal del gas transportador. El tiempo de retención es también directamente proporcional a la longitud de la columna, mientras que la resolución es proporcional a la raíz cuadrada de la longitud de la columna. Para las columnas rellenas, el caudal del gas transportador se expresa generalmente en ml por minuto a presión atmosférica y temperatura ambiente y se mide a la salida del detector con un caudalímetro mientras la columna está a la temperatura de trabajo. Para un caudal determinado, la velocidad lineal a través de una columna rellena es inversamente proporcional al cuadrado del diámetro de la columna. Para las columnas capilares habitualmente se emplea velocidad lineal en lugar de caudal.
- Un detector seleccionado de acuerdo a las características de la muestra. El detector debe mantenerse a una temperatura superior a la de la columna para impedir la condensación de las sustancias eluidas. Para los análisis cuantitativos los detectores deben brindar una respuesta que debe ser directamente proporcional a la cantidad de la sustancia presente en el detector para un intervalo amplio de concentraciones. El detector más comúnmente empleado es el de ionización a la llama pero también son empleados el de conductividad térmica, captura electrónica, nitrógeno-fósforo y espectrometría de masa.
Detector de ionización a la llama: poseen un intervalo lineal, amplio y es sensible a la mayoría de los compuestos orgánicos. La respuesta de los detectores depende de la estructura y la concentración del compuesto y del caudal del gas de combustión, del aire, del gas de compensación y del gas transportador. Cuando se emplean columnas rellenas el gas transportador puede ser helio o nitrógeno y cuando se emplean columnas capilares el gas transportador puede ser helio o hidrógeno, a menos que se especifique de otro modo en la monografía correspondiente.
Detector de conductividad térmica: posee un alambre a una temperatura determinada, colocado en la corriente del gas transportador. Mide la diferencia de conductividad térmica entre el gas transportador junto con la muestra y el gas transportador sólo cuando atraviesan el detector.
Este detector responde en forma uniforme a las sustancias volátiles cualquiera sea su estructura, sin embargo, es considerablemente menos sensible que el detector de ionización a la llama.
Detector de ionización a la llama alcalino: a veces llamado NP o detector de nitrógeno-fósforo, contiene una fuente termoónica, constituida por una sal de un metal alcalino o un elemento de vidrio que contiene rubidio u otro metal, que produce la ionización de compuestos con nitrógeno y fósforo orgánicos. Es un detector selectivo que muestra poca respuesta a los hidrocarburos.
Detector de captura electrónica: contiene una fuente de radiación ionizante. Presenta una respuesta sumamente alta con sustancias que contienen halógenos y grupos nitro pero pequeña con hidrocarburos. La sensibilidad aumenta con el número y peso atómico de los átomos de halógeno.
- Un registrador que recibe la señal del detector y calcula las respuestas de los picos e imprime el cromatograma con los parámetros del cromatograma y los picos. Los datos obtenidos pueden almacenarse y reprocesarse, con cambios en la integración y otras variables de cálculo según sea necesario.
Los datos también pueden recolectarse para ser medidos manualmente en registradores sencillos o en integradores que pueden calcular respuestas de picos o que producen cromatogramas con respuestas y alturas de los picos y permiten el almacenado de datos para un posible reprocesado.
Procedimiento - Acondicionar la columna, el inyector y el detector. Preparar las soluciones estándar y muestra según se especifica en la monografía correspondiente.
Calibrar el aparato con las soluciones estándar y determinar las cantidades a inyectar para obtener una respuesta apropiada.
Inyectar por separado las soluciones estándar y muestra, registrar los cromatogramas y medir las respuestas de los picos según se especifica en la monografía correspondiente.
Una fuente importante de error es la de irreproducibilidad en la cantidad de muestra inyectada, en particular cuando se hacen inyecciones manuales con una jeringa. Para reducir esta variabilidad, se agrega un estándar interno, compuesto que no interfiere en el cromatograma, en la misma concentración en las soluciones muestra y estándar. El cociente entre la respuesta de la sustancia ensayada y la respuesta del estándar interno se compara de un cromatograma a otro. El estándar interno debe ser sometido a todo el proceso de preparación de la muestra, para controlar además otros aspectos del análisis cuantitativo. Los inyectores automáticos mejoran la reproducibilidad de las inyecciones y reducen la necesidad del estándar interno.
A partir de los resultados obtenidos calcular el contenido de la o las sustancias a ensayar.
CROMATOGRAFIA DE LIQUIDOS DE ALTA EFICACIA
La Cromatografía de líquidos de alta eficacia, CLAE, (comúnmente llamada HPLC en inglés), es denominada también Cromatografía líquida de alta resolución o rendimiento. La Cromatografía líquida de alta eficacia tiene la ventaja de que las separaciones pueden tener lugar a temperatura ambiente para muchas sustancias. Por lo tanto, las sustancias no volátiles o térmicamente inestables, pueden cromatografiarse sin descomposición o necesidad de hacer derivados volátiles.
La afinidad de una sustancia por la fase estacionaria y, por consiguiente, su tiempo de retención en la columna, se controla variando la polaridad de la fase móvil mediante el agregado de un segundo y, a veces, un tercer o hasta un cuarto componente.
Aparato - Consta de:
- Un reservorio de fase móvil.
- Un sistema de bombeo que impulsa cantidades exactas de fase móvil desde el Reservorio de fase móvil a la columna mediante tuberías y uniones aptas para soportar altas presiones. Los sistemas modernos constan de una o varias bombas dosificadoras, controladas por computadora, que pueden programarse para variar la composición de la fase móvil cuando se trabaja con gradiente o para mezclar los componentes de la fase móvil cuando se trabaja en condiciones isocráticas. Generalmente se trabaja con gradiente cuando la muestra es muy compleja o contiene sustancias que difieren mucho en su factor de capacidad.
Las bombas pueden dar origen a caudales de hasta 10 ml por minuto y generar presiones de hasta 6.000 psi. Sin embargo, los caudales típicos son de 1 a 2 ml por minuto, con presiones no mayores a 2.000 ó 2.500 psi. Las bombas empleadas para el análisis cuantitativo son de material resistente tanto al ataque químico como al ataque mecánico y son capaces de entregar la fase móvil a una velocidad constante, con fluctuaciones mínimas, durante períodos prolongados.
- Un sistema de inyección empleado para introducir la muestra.
- Una columna donde se produce la separación efectiva de los componentes de la muestra inyectada.
Las fases estacionarias para la cromatografía de líquidos en fase reversa constan de una fase orgánica químicamente unida a sílice u otros materiales. Las partículas son generalmente de 3 a 10 mm de diámetro pero los tamaños pueden llegar hasta 50 mm o más para las columnas preparativas. Las partículas recubiertas con una capa delgada de fase orgánica tienen una baja resistencia a la transferencia de masa y, por lo tanto, se obtiene una transferencia rápida de las sustancias entre la fase estacionaria y la fase móvil. La polaridad de la fase estacionaria depende de la polaridad de sus grupos funcionales que van desde el octadecilsilano relativamente no polar a grupos nitrilo muy polares.
Por lo general, las columnas empleadas para las separaciones analíticas tienen diámetros internos de 2 a 5 mm; para la cromatografía preparativa se emplean columnas de diámetro mayor. En algunos casos las columnas pueden calentarse para mejorar la eficiencia durante la separación, pero rara vez se las emplea a temperaturas por encima de los 60°C, debido a la potencial degradación de la fase estacionaria o a la volatilidad de la fase móvil. Las columnas se emplean a temperatura ambiente, a menos que se especifique de otro modo en la monografía correspondiente.
- Un detector. Se emplean generalmente:
Detector espectrofotométrico: consta de una celda de flujo colocada a la salida de la columna. Un haz de radiación ultravioleta pasa a través de la celda de flujo en forma perpendicular a la dirección del flujo de la fase móvil e incide en el fotodetector. A medida que las sustancias eluyen de la columna, pasan a través de la celda y absorben la radiación, lo que da lugar a cambios cuantificables en el nivel de energía. Este detector es el más empleado.
Existen detectores de longitud de onda fija, variable y múltiple. Los detectores de longitud de onda fija operan a una sola longitud de onda, en general 254 nm, emitida por una lámpara de mercurio de baja presión. Los detectores de longitud de onda variable contienen una frente continua, como una lámpara de deuterio o de xenón de alta presión y un monocromador o un filtro de interferencia que generan una radiación monocromática a una longitud de onda seleccionada por el analista. Los detectores de longitud de onda variable pueden programarse para variar la longitud de onda durante el análisis. Los detectores de longitud de onda múltiple miden la absorbancia a dos o más longitudes de onda simultáneamente.
Detectores por arreglo de diodos: el haz de luz continua atraviesa la celda. Luego la radiación es resuelta en sus longitudes de onda constitutivas que son detectadas individualmente mediante el arreglo de diodos. Estos detectores adquieren los datos de absorbancia sobre el intervalo total UV-visible y, por lo tanto, proveen al analista varias longitudes de onda seleccionadas y espectros de los picos eluidos. Los arreglos de diodos tienen, por lo general, menor relación señal-ruido que los detectores de longitud de onda fija o variable y, por lo tanto, son menos apropiados para el análisis de sustancias presentes en bajas concentraciones.
Detectores de índice de refracción: miden la diferencia entre el índice de refracción de la fase móvil sola y el de la fase móvil que contiene las sustancias cromatografiadas según eluyan de la columna. Se emplean para detectar sustancias que no absorben radiación ultravioleta, pero son menos sensibles que los detectores ultravioleta. Son sensibles a pequeños cambios en la composición del solvente, el caudal y la temperatura, por ello se emplea una celda de referencia que contiene la fase móvil para obtener una línea de base satisfactoria.
Detectores fluorométricos: son sensibles a las sustancias fluorescentes o que puedan convertirse en derivados fluorescentes, ya sea mediante la transformación química de la sustancia o acoplando reactivos fluorescentes en grupos funcionales específicos.
Detectores electroquímicos, potenciométricos, voltamétricos o polarográficos: son útiles para la cuantificación de sustancias que pueden oxidarse o reducirse en un electrodo de trabajo. Estos detectores son selectivos, sensibles y confiables pero las fases móviles deben estar libres de oxígeno disuelto o iones metálicos oxidantes. Debe emplearse una bomba sin pulso y el pH, la fuerza iónica y la temperatura de la fase móvil deben permanecer constantes. Los electrodos de trabajo son propensos a la contaminación por los productos de reacción con las consiguientes variaciones en las respuestas. Los detectores electroquímicos con electrodos de pasta de carbono pueden emplearse ventajosamente para medir cantidades muy pequeñas, del orden de los ng de sustancias fácilmente oxidables en particular fenoles y catecoles.
-Un registrador que recibe la información del detector y realiza la impresión del cromatograrna. Los dispositivos modernos almacenan la señal de salida del detector permitiendo reprocesar el cromatograma luego de cambiar las variables de integración. También pueden emplearse para programar el cromatógrafo controlando la mayoría de las variables y automatizando el proceso.
Procedimiento - La composición de la fase móvil influye significativamente en la resolución de las sustancias en la muestra. [NOTA: deben emplearse solventes de grado HPLC y reactivos de alta pureza].
Equilibrar la columna con la fase móvil y preparar las soluciones estándar y muestra según se especifique en la monografía correspondiente. Las soluciones deben estar exentas de partículas sólidas.
Calibrar el aparato con las soluciones estándar y determinar las cantidades a inyectar para obtener una respuesta apropiada. Determinar la reproducibilidad del sistema cromatográfico realizando inyecciones repetidas y, si fuera necesario, calcular el número de platos teóricos.
Inyectar por separado las soluciones estándar y muestra, registrar los cromatogramas y medir las respuestas de los picos según se especifique en la monografía correspondiente.
A partir de los valores obtenidos calcular el contenido de la o las sustancias a ensayar.
Los sistemas CLAE se calibran para el análisis cuantitativo con Sustancias de referencia. Los métodos generalmente empleados son los de estándar externo y estándar interno. En general se obtienen resultados confiables por los sistemas de estándar externo, especialmente cuando se emplean inyectores automáticos. Este método compara directamente la respuesta obtenida cromatografiando separadamente soluciones estándar y muestra. En otros casos se obtienen mejores resultados empleando el sistema de estándar interno. En este caso se agrega una cantidad conocida de una sustancia no interferente, el estándar interno, a las soluciones muestra y estándar. Luego se compara la relación de respuesta estándar/estándar interno y muestra/estándar interno.
CROMATOGRAFIA DE EXCLUSION
En la Cromatografía de exclusión las sustancias presentes en la muestra se separan de acuerdo a su tamaño. Los compuestos cuyas dimensiones sean mayores que el tamaño de poro de la fase estacionaria atraviesan la columna sin ser retenidos y eluyen al volumen de exclusión VO (volumen muerto). Los compuestos cuyas dimensiones sean menores que el tamaño de poro de la fase estacionaria se introducen en los poros, quedan retenidos por más tiempo y eluyen al volumen total de permeación VT (cuanto menor sea dicho tamaño más tiempo quedan retenidos en los poros y viceversa).
La Cromatografía de exclusión se puede dividir en Cromatografía de permeación de geles que emplea fases móviles orgánicas no polares y rellenos hidrofílicos y Cromatografía por filtración de geles que emplea fases móviles acuosas y rellenos hidrofóbicos.
Aparato - Emplear el cromatógrafo descripto en Cromatografía de líquidos de alta eficacia.
- Columna. [NOTA: si fuera necesario, controlar la temperatura de la columna].
El material de relleno puede ser un soporte blando, como por ej., un gel o un soporte rígido, cono por ej., vidrio, sílica o un polímero orgánico entrecruzado compatible con la fase móvil.
- Detector. Generalmente los detectores se basan en propiedades luminiscentes, refractarias o fotométricas (ver Detectores en Cromatografía de líquidos de alta eficacia). [NOTA: si fuera necesario, se puede conectar a un colector automático de fracciones].
Procedimiento - Rellenar la columna según se especifica en la monografía correspondiente. Las características de elución de un compuesto en un sistema cromatográfico determinado se puede describir por el coeficiente de distribución, KD, el cual se calcula por la fórmula siguiente:
( VI - VO)/( VT - VO)
en la cual VO es el volumen de retención para la sustancia no retenida, VT es el volumen de retención para la sustancia que tiene acceso total a todos los poros del material de relleno y VI es el volumen de retención para la sustancia a ensayar. Medir cada volumen de retención desde el momento de la aplicación hasta el momento de cada pico máximo en particular.
Determinación de la composición relativa de cada componente en la mezcla - Proceder para la separación según se especifica en la monografía correspondiente. Medir las respuestas de los picos principales. Si todos los componentes dada muestra poseen propiedades fisicoquímicas equivalentes, como por ej., la absortividad, calcular la cantidad relativa de cada componente dividiendo la respuesta del pico respectivo por la suma de las respuestas de los picos de todas las sustancias de ensayo. Si los componentes de la muestra no poseen propiedades fisicoquímicas equivalentes calcular el contenido empleando curvas de calibración obtenidas según se especifica en la monografía correspondiente.
Determinación de pesos moleculares - Determinar los pesos moleculares de los componentes de la muestra comparando con los estándar de calibración especificados en las monografías correspondientes. Graficar los volúmenes de retención de los estándar de calibración en función del logaritmo de sus pesos moleculares. Trazar la línea recta que mejor se ajuste a los puntos graficados dentro de los límites de exclusión total y de permeación total. Los pesos moleculares de los componentes de la muestra se estiman a partir de la curva de calibración.
Determinación de la distribución de pesos moleculares en polímeros - Proceder según se especifica en las monografías correspondientes.
INTERPRETACION DE LOS CROMATOGRAMAS
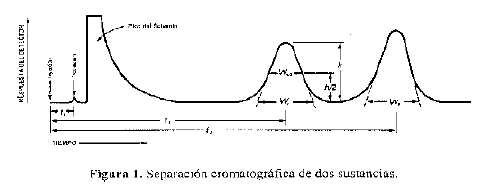
En la Figura 1 se representa una separación cromatográfica típica de dos sustancias; 1 y 2, donde t1 y t2 son los tiempos de retención, respectivamente; h, h/2 y Wh/2 son la altura, la mitad de la altura y el ancho a la mitad de la altura, respectivamente, para el pico 1; W1 y W2 son los anchos de los picos 1 y 2 en la línea de base, respectivamente.
La coincidencia de los tiempos de retención de una muestra y una Sustancia de referencia en un único sistema cromatográfico puede emplearse como una característica en la construcción de un perfil de identidad, pero es insuficiente para establecer la identidad. Los tiempos de retención absolutos de un compuesto dado varían de un cromatograma al próximo.
[NOTA: en las siguientes fórmulas, tanto los volúmenes de retención como las separaciones lineales son directamente proporcionales al tiempo de retención y pueden sustituirse en las fórmulas]. Normalmente las comparaciones se hacen en función de la retención relativa, a, que se calcula por la fórmula siguiente:
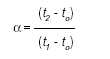
en la cual t2 y t1 son los tiempos de retención de la muestra y del estándar respectivamente, medidos desde el punto de inyección, en condiciones experimentales idénticas en la misma columna y tO es el tiempo de retención de una sustancia no retenida, como por ej., metano en el caso de la Cromatografía de gases.
El número de platos teóricos N, es una medida de la eficiencia de la columna. Para los picos gaussianos, se calcula por la fórmula siguiente:
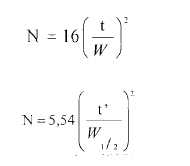
en la cual t es el tiempo de retención de la sustancia, W es el ancho del pico en su base, obtenido al extrapolar los lados del pico con la línea de base y t’ es la diferencia entre el tiempo de retención de la sustancia y el tiempo de elución de una sustancia que no es retenida (tiempo muerto). El ancho máximo a media altura, W1/2 , es obtenido directamente por integradores electrónicos.
El valor de N depende de la sustancia cromatografiada, así como de las condiciones operativas, como por ej., la fase móvil, el caudal del gas transportador, la temperatura, la calidad y uniformidad de la fase estacionaria y, para las columnas capilares, el espesor de la película de fase estacionaria, el diámetro y la longitud de la columna.
Para la separación de dos sustancias en una mezcla, la resolución, R, es determinada por la fórmula siguiente:
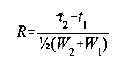
en la cual t2 y t1 son los tiempos de retención de los dos componentes y W2 y W1 son los anchos de la base de los picos correspondientes, obtenidos al extrapolar los lados con la línea de base.
La respuesta y la altura del pico son generalmente proporcionales a la cantidad de sustancia eluida. En general se miden las respuestas de los picos, sin embargo, la medición de las alturas pueden ser más exactas que la respuesta cuando se consideran picos parcialmente superpuestos.
El ensayo de pureza cromatográfica de una materia prima se basa en la determinación de picos de impurezas y se expresa como un porcentaje de la respuesta del pico de la sustancia. Es preferible, sin embargo, comparar los picos de impurezas con el cromatograma de un estándar a una concentración similar. El estándar puede ser la misma sustancia a un nivel que corresponde, como por ej., 0,5% de impureza, o en el caso de las impurezas tóxicas o de impurezas indicadoras, un estándar de la impureza misma.
El factor de capacidad k’, está relacionado con el coeficiente de distribución de la sustancia a ensayar entre ambas fases. El factor de capacidad depende de la naturaleza química de la sustancia; del área, naturaleza y cantidad de fase estacionaria; de la temperatura de la columna y del caudal de la fase móvil. Cada sustancia tiene un factor de capacidad característico para un conjunto específico de condiciones experimentales. La separación cromatográfica sólo se produce si las sustancias involucradas poseen diferentes factores de capacidad. El factor de capacidad se calcula por la fórmula siguiente:
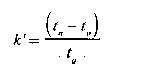
en la cual tn es el tiempo requerido para que la sustancia a ensayar eluya a través de la columna (tiempo de retención) y to es el tiempo de elución de una sustancia que no es retenida (tiempo muerto).
APTITUD DEL SISTEMA
Los ensayos de aptitud del sistema forman parte de los métodos cromatográficos líquido y de gases. Se emplean para comprobar que la resolución y la reproducibilidad del sistema cromatográfico son aptas para realizar el ensayo. Para estos ensayos se considera que el equipo, las operaciones analíticas y las muestras a ensayar constituyen un sistema único que puede evaluarse corno tal.
Los parámetros de aptitud del sistema se determinan para el pico de la sustancia, a menos que se especifique de otro modo en la monografía correspondiente.
Si es necesario, pueden realizarse ajustes en las condiciones operativas para cumplir con los requisitos de aptitud del sistema, entre ellos, las proporciones de los componentes de la fase móvil y el caudal.
La resolución R, es una función de la eficiencia de la columna N, y se especifica para asegurar que las Sustancias que eluyan muy cercanas se resuelvan entre sí y para asegurar que los estándares internos se resuelvan de las sustancias a ensayar. La eficiencia de la columna puede especificarse también como un requisito de aptitud del sistema, especialmente si hay un solo pico de interés en el cromatograma, sin embargo, el valor aislado de eficiencia no puede asegurar la resolución para el sistema en estudio. La eficiencia de la columna es una medida de la agudeza de los picos, importante para detectar componentes en baja concentración.
Para evaluar si se cumplen los requisitos de aptitud del sistema se realizan inyecciones repetidas de la Preparación estándar empleada en la Valoración u otra Solución estándar. A menos que se especifique de otro modo en la monografía correspondiente, para calcular la desviación estándar relativa, SR, se emplean los datos de cinco inyecciones repetidas del estándar si el requisito es 2,0% o menor, y seis inyecciones repetidas si el requisito de la desviación estándar relativa es mayor de 2,0%. La desviación estándar relativa SR, se calcula según la fórmula siguiente:

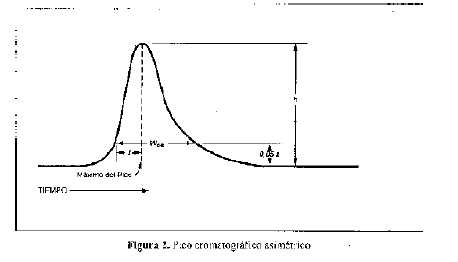
El factor de asimetría F, una medida de la simetría del pico, es 1 para los picos perfectamente simétricos y su valor aumenta a medida que la asimetría es más pronunciada (ver Figura 2). En algunos casos, pueden observarse valores menores de la unidad. Como consecuencia de la asimetría del pico, la integración y la precisión se tornan menos confiables.
EI factor de asimetría se calcula por la fórmula siguiente:
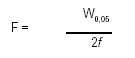
en la cual W0,05 es el ancho del pico al 5% de altura y f es la distancia entre el borde simétrico del pico y el centro del mismo medida al 5% de la altura.
Estos datos se obtienen a partir de Inyecciones repetidas del estándar u otras soluciones según se especifique en la monografía correspondiente.