300. ELECTROFORESIS
Las partículas cargadas, disueltas o dispersas en una solución electrolítica migran hacia el electrodo de polaridad opuesta, bajo la acción de un campo eléctrico.
En la electroforesis en gel, el desplazamiento de las partículas se retarda por las interacciones con la matriz de gel que constituye el medio de migración y se comporta como un tamiz molecular. Las interacciones entre las fuerzas eléctricas y la tamización molecular dan lugar a una velocidad de migración diferencial según el tamaño, la forma y la carga de las partículas. Las diferentes macromoléculas presentes en una mezcla migran a diferentes velocidades durante el proceso electroforético, debido a sus propiedades fisicoquímicas, separándose en fracciones concretas.
Las separaciones electroforéticas se pueden realizar sin soporte, como por ej., en la electroforesis capilar en solución libre o en medios estabilizantes como placas de capa delgada, películas y geles.
ELECTROFORESIS DE LIBRE MIGRACION O FRONTAL
Este método se emplea fundamentalmente para determinar movilidades electroforéticas experimentalmente y se caracteriza por brindar medidas directas y reproducibles. Se aplica principalmente a sustancias de alto peso molecular y poco difundibles. Las bandas se localizan en principio mediante un método físico como refractometría o conductimetría. Luego de la aplicación de un campo eléctrico definido durante un tiempo exactamente medido, se observa la ubicación de las nuevas bandas obtenidas. Las condiciones operativas deben ser tales que permitan obtener tantas bandas como componentes haya en la muestra.
ELECTROFORESIS SOBRE UN SOPORTE O ELECTROFORESIS DE ZONA
Este método requiere el empleo de cantidades de muestra reducidas.
Existen diferentes tipos de soportes, como papel, gel de agar, acetato de celulosa, almidón, agarosa, metacrilamida y gel mixto. La naturaleza del soporte introduce numerosos factores que modifican la movilidad:
a - debido a las sinuosidades de los canalículos del soporte, la distancia recorrida aparentemente es menor que la real;
b - algunos soportes no son eléctricamente neutros; esto provoca, en muchas ocasiones, un flujo electroendosmótico importante;
c - efectos de calentamiento debidos al efecto joule pueden provocar evaporación del solvente desde el soporte y por capilaridad, ocurriendo un movimiento de la solución desde los extremos hacia el centro. Por lo tanto, la fuerza iónica, tiende a aumentar progresivamente.
La velocidad de migración depende fundamentalmente de cuatro factores: movilidad de la partícula cargada, flujo electroendosmótico, flujo de evaporación y campo eléctrico. Por lo tanto, es necesario trabajar en condiciones experimentales claramente definidas y emplear, siempre que sea posible, Sustancias de referencia.
Aparato - Consta de:
- Generador de corriente contínua, cuyo voltaje se pueda controlar y estabilizar.
- Cubeta electroforética. Generalmente son rectangulares, de vidrio o plástico rígido, con dos compartimentos separados, el anódico y el catódico, que contienen la solución de electrolito. En cada compartimento se introduce un electrodo de, por ej., platino o grafito; los cuales se conectan por medio de un circuito convenientemente aislado a los correspondientes terminales del Generador de corriente contínua para constituir el ánodo y el cátodo. El nivel de líquido en ambos compartimentos se debe mantener igualado para prevenir efecto sifón.
La cubeta electroforética se cierra herméticamente para mantener un ambiente saturado de humedad durante todo el proceso y reducir la evaporación de solvente. Se puede emplear un dispositivo de seguridad que corte el paso de corriente eléctrica cuando se levanta la tapa. Si la potencia eléctrica que pasa a través del soporte excede los 10 W, es recomendable refrigerar el soporte.
- Dispositivo portasoportes:
Para electroforesis en tiras. La tira soporte, previamente humedecida con la solución conductora y con los extremos sumergidos en los correspondientes compartimentos electródicos, se fija y se tensa sobre un portasoporte apropiado, diseñado para prevenir la difusión del electrolito conductor de la corriente eléctrica, como un bastidor horizontal, un caballete en V invertida o una superficie uniforme con puntos de contacto a intervalos apropiados.
Para electroforesis en gel. El dispositivo consta esencialmente de una placa de vidrio (por ej., un portaobjetos de microscopio) sobre la que se deposita, en la totalidad de su superficie, una capa firmemente adherida de gel de espesor uniforme. La conexión entre el gel y la solución conductora se realiza de varios modos, dependiendo del tipo de aparato empleado. Se deben tomar precauciones para evitar la condensación de humedad o el desecado de la capa sólida.
- Dispositivo de medida o medio de detección.
Procedimiento - Transferir la solución del electrolito a los compartimentos electródicos. Colocar el soporte apropiadamente impregnado con el electrolito en la cubeta según las condiciones indicadas para el tipo de aparato empleado. Localizar la zona de aplicación y aplicar la muestra. Aplicar la corriente eléctrica durante el tiempo especificado. Luego de desconectar la corriente, retirar el soporte de la cubeta electroforética, secar y revelar.
ELECTROFORESIS SOBRE GEL CILINDRICO DE POLIACRILAMIDA
En la electroforesis sobre gel cilíndrico de poliacrilamida, la fase estacionaria está constituida por un gel preparado con una mezcla de acrilamida y N, N’- metilenobisacrilamida; los geles se preparan en tubos, generalmente de 0,5 cm de diámetro interno y 7,5 cm de longitud; en cada tubo se aplica una sola muestra.
Aparato - Consta de dos compartimentos destinados a las soluciones reguladoras, fabricados con un material apropiado como polimetacrilato de metilo, dispuestos en posición vertical uno arriba del otro. Cada compartimento está provisto de un electrodo de platino que se conecta con un generador de corriente contínua que permite trabajar a intensidad constante o voltaje constante. Para los geles cilíndricos, el compartimento superior está provisto en su base de varios dispositivos para los tubos situados equidistantes al electrodo.
Procedimiento - [NOTA: se recomienda desgasificar las soluciones antes de la polimerización y emplear el gel inmediatamente después de su preparación]. Preparar la mezcla de geles según se especifica en la monografía correspondiente. Verter la mezcla de gel en tubos apropiados, tapados en la parte inferior, igualar el nivel de gel en cada uno de los tubos y rellenar hasta 1 cm del borde superior. Evitar que queden burbujas de aire atrapadas en el interior de los tubos. Cubrir la mezcla de gel con una capa de agua para evitar el contacto con el aire y dejar reposar para que ocurra la gelificación. Por lo general, la formación del gel requiere aproximadamente 30 minutos y se completa cuando se produce una interfase nítida entre el gel y la capa de agua. Eliminar la capa de agua, rellenar el compartimento inferior con la solución reguladora indicada en la monografía correspondiente y destapar los tubos. Introducir los tubos en los dispositivos del compartimento superior y ajustarlos de modo que la parte inferior de los tubos se encuentre inmersa en la solución reguladora del compartimento inferior. Rellenar los tubos cuidadosamente con la solución reguladora especificada. Preparar la solución muestra y la solución de referencia con el identificador especificado y aumentar la densidad de estas soluciones mediante el agregado de sacarosa. Aplicar ambas soluciones en la superficie del gel, empleando un tubo diferente para cada solución. Agregar la misma solución reguladora en el compartimento superior. Conectar los electrodos al generador de corriente y desarrollar la electroforesis a la temperatura y el voltaje o intensidad de corriente constantes especificados en la monografía correspondiente.
ELECTROFORESIS EN GEL DE POLIACRILAMIDA CON DODECIL SULFATO DE SODIO (SDS-PAGE)
La electroforesis en gel de poliacrilamida se emplea con fines de caracterización cualitativa de proteínas en preparaciones biológicas, control de pureza y determinaciones cuantitativas.
La electroforesis analítica en gel es un método apropiado para identificar y controlar la homogeneidad de las proteínas en productos farmacéuticos. También se emplea para estimar los pesos moleculares de subunidades proteicas y para determinar las subunidades que componen las proteínas purificadas.
Características de los geles de poliacrilamida
Las propiedades de tamización de los geles de poliacrilamida se deben a su particular estructura, una red tridimensional de fibras y poros obtenida mediante enlaces cruzados de unidades de bisacrilamida bifuncionales con cadenas adyacentes de poliacrilamida. La polimerización se cataliza a través de un generador de radicales libres constituido por persulfato de amonio y tetrametiletilendiamina.
Si se aumenta la concentración de acrilamida del gel, disminuye su tamaño de poro efectivo. Este se define, desde el punto de vista operativo, por sus propiedades de tamización molecular; es decir, la resistencia con la que se opone a la migración de macromoléculas. Las concentraciones de acrilamida que se pueden emplear se encuentran dentro de ciertos límites ya que a altas concentraciones los geles se rompen con mayor facilidad y su manejo es más dificultoso.
Cuando el tamaño de poro, disminuye, la velocidad de migración de la molécula a través del gel decrece. La resolución para un producto determinado se puede optimizar ajustando el tamaño de poro del gel o modificando la concentración de acrilamida. Por lo tanto, las características físicas de un gel determinado dependen de su contenido de acrilamida y de bisacrilamida.
Además de la composición del gel, el estado de la molécula constituye un factor importante que afecta la movilidad electroforética. Para las proteínas, la movilidad electroforética depende del pK de los grupos cargados y del tamaño de la molécula; siendo afectada también por la naturaleza, concentración y pH de la solución reguladora, la temperatura, la intensidad del campo eléctrico aplicado y la naturaleza del material del soporte.
Electroforésis en gel de poliacrilamida con desnaturalización
Este método se emplea para el análisis de monómeros polipeptídicos de peso molecular comprendido entre 14.000 y 100.000.
La electroforesis en gel de poliacrilamida con desnaturalización, empleando dodecilsulfato de sodio (SDS-PAGE), es la técnica electroforética más empleada para la evaluación de la calidad de productos proteicos. De modo general, la electroforesis analítica de proteínas se realiza en geles de poliacrilamida en condiciones en las que se asegure la disociación de las proteínas en sus subunidades polipeptídicas individuales, limitándose los procesos de agregación. Se emplea frecuentemente para disociar las proteínas antes de la aplicación en el gel, el dodecilsulfato de sodio (SDS), un detergente aniónico fuerte, en combinación con calor. Los polipéptidos desnaturalizados se unen al SDS, adquieren carga negativa y presentan una relación carga/masa constante, independientemente del tipo de proteína considerada. La cantidad de SDS es casi siempre proporcional al peso molecular del polipéptido y es independiente de su secuencia ya que los complejos SDS-polipéptido migran a través de los geles de poliacrilamida con movilidades que dependen del tamaño del polipéptido.
Las movilidades electroforéticas de los complejos SDS-polipéptido resultantes presentan siempre la misma relación funcional con los pesos moleculares. La migración de los complejos SDS-polipéptido se efectúa hacia el ánodo, a una velocidad superior para los complejos de peso molecular más bajo que para aquéllos con peso molecular alto. De este modo, es posible estimar el peso molecular de una proteína a partir de su movilidad relativa en un método SDS-PAGE calibrado, siendo la presencia de una única banda en dicho gel un criterio de pureza.
Las eventuales modificaciones del esqueleto polipéptidico, como por ej., una O- o N-glicosilación, tiene un impacto significativo sobre el peso molecular aparente de la proteína ya que el SDS no se une del mismo modo a los grupos glucídicos que a los grupos polipéptidicos, por lo que en este caso no se mantiene constante la relación carga/masa.
Condiciones reductoras - La asociación de las subunidades polipéptidicas y la estructura tridimensional de las proteínas se mantiene fundamentalmente por la existencia de enlaces disulfuro. Uno de los objetivos de la separación SDS-PACE en condiciones reductoras es la ruptura de esta estructura por reducción de los enlaces disulfuro. La desnaturalización y disociación completa de las proteínas por tratamiento con 2- mercaptoetanol o ditiotreitol (DTT) da lugar a un desdoblamiento de la cadena polipéptidica, seguido de la formación de un complejo con el SDS. En estas condiciones, el peso molecular de las subunidades polipéptidicas se puede calcular por regresión lineal en presencia de patrones con pesos moleculares apropiados.
Condiciones no reductoras - Para algunos análisis no es aconsejable la disociación completa de la proteína en subunidades peptídicas. Si no se emplean agentes reductores como el 2-mercaptoetanol o DTT, los enlaces covalentes disulfuro permanecen intactos manteniéndose la forma oligomérica de la proteína. Los complejos SDS-proteína oligomérica migran más lentamente que sus subunidades SDSpolipéptido. Además, las proteínas no reducidas no se pueden saturar completamente con SDS y por lo tanto, no pueden unirse al detergente en una proporción de masa constante. Esto hace que la determinación del peso molecular de estas moléculas por SDS-PACE sea más difícil que los análisis de los polipéptidos totalmente desnaturalizados, ya que es necesario que tanto las proteínas patrón como las proteínas de la muestra presenten configuraciones similares para que se puedan comparar. Sin embargo, la obtención en el gel de una sola banda coloreada es un criterio de pureza.
CARACTERISTICAS DE LA ELECTROFORESIS EN GEL CON SISTEMA REGULADOR DISCONTINUO
El método electroforético más usado para la caracterización de mezclas complejas de proteínas se basa en el empleo de un sistema regulador discontinuo consistente en dos geles contiguos pero distintos: un gel inferior, llamado gel de separación o de resolución, y otro superior, gel de apilamiento. Estos dos geles presentan porosidad, pH y fuerzas iónicas diferentes. Además se emplean iones con diferentes movilidades iónicas en el gel y en la solución reguladora. La discontinuidad del sistema provoca una concentración de las muestras de mayor tamaño en el gel de apilamiento y por lo tanto se mejora la resolución. Cuando se aplica la corriente eléctrica, se desarrolla un gradiente de potencial negativo a través de la solución muestra que conduce a las proteínas hacia el gel de apilamiento. Los iones glicinato contenidos en la solución reguladora empujan a las proteínas hacia el gel de apilamiento. Se forma rápidamente una zona de frente móvil cuya cabecera está constituida por iones cloruro de alta movilidad, seguidos de iones glicinato más lentos en la cola. Se produce un gradiente de alto voltaje localizado entre los frentes iónicos de cabeza y cola, provocando que los complejos SDS-proteína se concentren en una zona muy estrecha (apilamiento) y migren entre las fases de cloruro y glicinato. Independientemente del volumen de muestra aplicado, el conjunto de complejos SDS-proteína se condensa en una región muy estrecha y penetran en el gel de separación en forma de banda estrecha, bien definida y de alta densidad proteica. El gel de apilamiento, de tamaño de poro, grande, no retarda la migración de la mayoría de las proteínas y actúa principalmente como medio anticonvectivo. En la interfase de ambos geles, las proteínas experimentan un incremento brusco de retardo electroforético debido al menor tamaño de poro del gel de resolución. Una vez que se encuentran en este gel, las proteínas continúan avanzando lentamente por el efecto de tamización molecular que ejerce la matriz. Los iones glicinato migran por delante de las proteínas, por lo que éstas se mueven en un medio de pH uniforme formado por el tris(hidroximetil)aminometano y la glicina. La tamización molecular hace que la separación de los complejos SDS-polipéptido se base en sus correspondientes pesos moleculares.
PREPARACION DE GELES DE POLIACRILAMIDA SDS VERTICALES CON SISTEMA REGULADOR DISCONTINUO
Preparación de los geles
En un gel de poliacrilamida con sistema regulador discontinuo, se recomienda verter el gel de resolución, dejar polimerizar y a continuación verter el gel de apilamiento ya que la constitución de los dos geles de acrilamida-bisacrilamida es diferente.
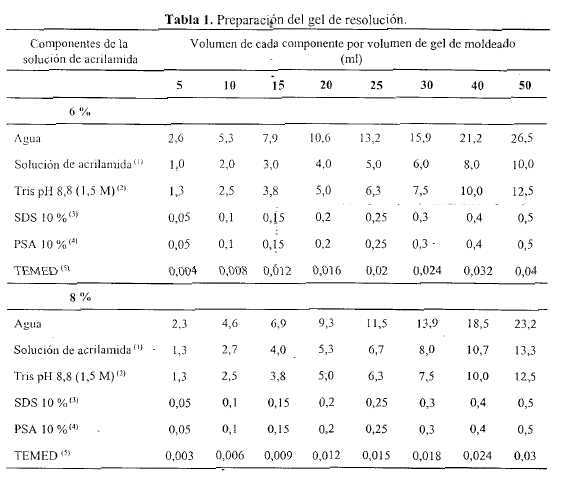
Preparación del gel de resolución - En un erlenmeyer, preparar el volumen apropiado de la solución de acrilamida que contenga la concentración deseada para el gel de resolución, empleando los valores indicados en la Tabla 1. Mezclar los componentes en el orden indicado. Antes del agregado de la solución de persulfato de amonio y del tetrametiletilendiamina (TEMED), filtrar la solución, si fuera necesario, empleando vacío, a través de una membrana de acetato de celulosa (de 0,45 mm de diámetro); mantener la solución bajo vacío por rotación de la unidad de filtración hasta que no se formen más burbujas. Agregar las cantidades apropiadas de solución de persulfato de amonio y de TEMED indicadas en la Tabla 1, agitar y verter rápidamente en el espacio de separación entre las dos placas de vidrio del molde. Dejar espacio suficiente para el gel de apilamiento (la longitud de un diente del peine más 1 cm). Empleando una pipeta de vidrio de punta larga, recubrir la solución con isobutanol saturado con agua. Dejar el gel en posición vertical a temperatura ambiente para que se produzca la polimerización.
Preparación del gel de apilamiento - Luego de la polimerización completa del gel de resolución (aproximadamente 30 minutos), retirar la capa superior de isobutanol y lavar varias veces con agua la parte superior del gel para eliminar la capa de isobutanol y la posible acrilamida no polimerizada. Eliminar la mayor cantidad posible de líquido de la superficie del gel eliminando el resto de agua con la ayuda de un papel de filtro.
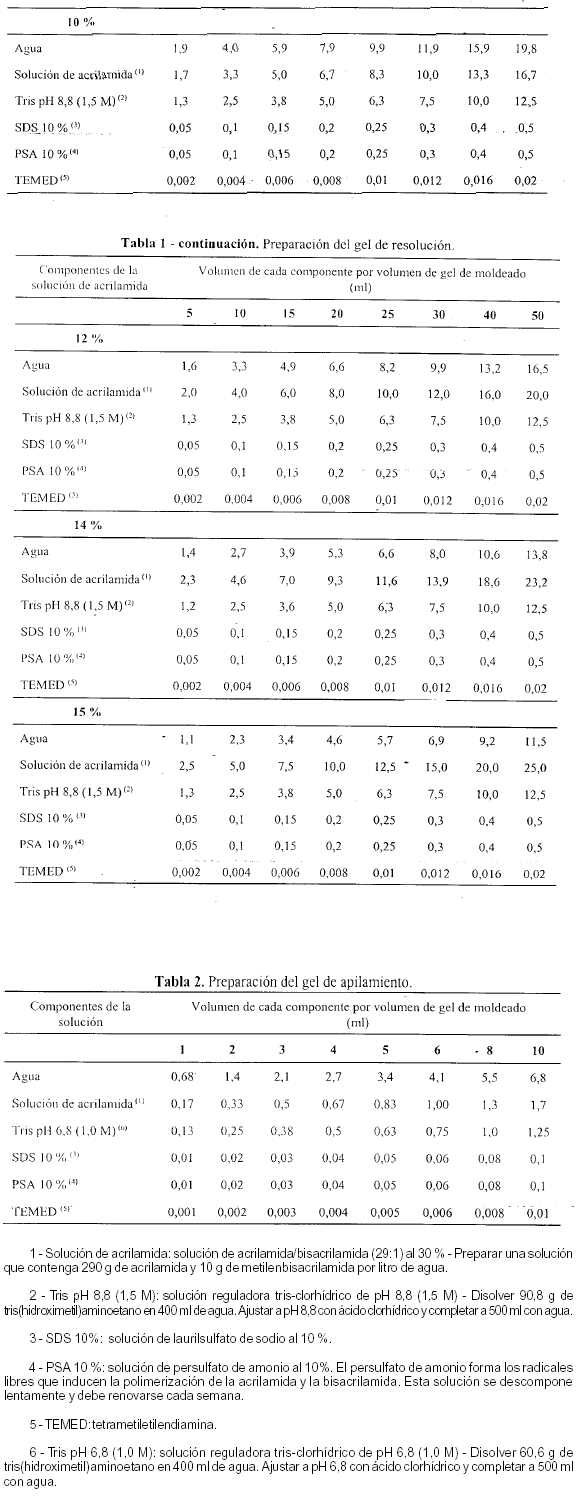
En un erlenmeyer, preparar un volumen apropiado de solución que contenga la concentración requerida para el gel de resolución, según se indica en la Tabla 2. Mezclar los componentes en el orden indicado. Antes del agregado de la solución de persulfato de amonio y del tetrametiletilendiamina, filtrar la solución, si fuera necesario, empleando vacío, a través de una membrana de acetato de celulosa (de 0,45 mm de diámetro); mantener la solución bajo vacío por rotación de la unidad de filtración hasta que no se formen más burbujas. Agregar las cantidades apropiadas de solución de persulfato de amonio y de TEMED, indicadas en la Tabla 2, agitar y verter rápidamente en el espacio de separación entre las dos placas de vidrio del molde, directamente sobre la superficie del gel de resolución polimerizado. De inmediato, insertar un peine limpio de politetrafluoroetileno en la solución del gel de apilamiento, evitando la formación de burbujas de aire. Agregar más solución del gel de apilamiento hasta rellenar completamente los espacios del peine. Colocar el gel en posición vertical y dejar polimerizar a temperatura ambiente.
Montaje del gel en el equipo de electroforesis y separación electroforética
Solución reguladora de desarrollo SDS-PAGE - Disolver en agua 151,4 g de tris(hidroximetil)aminoetano, 721,0 g de glicina y 50,0 g de lauril sulfato de sodio, y completar a 5 litros con agua. Inmediatamente antes del uso, diluir 10 veces su volumen con agua y mezclar. El pH de esta solución debe estar comprendido entre 8,1 y 8,8.
Procedimiento - Una vez completada la polimerización (aproximadamente 30 minutos), retirar cuidadosamente el peine de politetrafluoroetileno y lavar los pocitos, de inmediato, con agua o Solución reguladora de desarrollo SDS-PAGE para eliminar la posible acrilainida no polimerizada. Recortar los dientes del gel de apilamiento, si fuera necesario, con una aguja hipodérmica roma fijada a una jeringa. Quitar las pinzas de uno de los lados cortos, retirar el tubo con precaución y volver a colocarlas. Proceder del mismo modo en el otro. Retirar el tubo de la parte inferior del gel, montar el gel en el aparato de electroforesis y agregar las soluciones reguladoras en los compartimentos superior e inferior. Eliminar las burbujas que se formen en la parte inferior del gel, entre las dos placas de vidrio; preferiblemente efectuar este procedimiento con una aguja hipodérmica doblada fijada a una jeringa. [NOTA: nunca realizar un predesarrollo sin muestras ya que se destruye la discontinuidad de los sistemas reguladores]. Antes de aplicar las muestras, lavar cuidadosamente la ranura con Solución reguladora de desarrollo SDS-PAGE. Preparar la solución muestra y la solución de referencia en el medio recomendado y proceder según se especifica en la monografía correspondiente. Aplicar el volumen apropiado de cada solución en los pocitos del gel de apilamiento y desarrollar la electroforesis. En ocasiones resulta necesario modificar el tiempo y la relación intensidad/voltaje, para obtener una separación óptima. Comprobar que el frente del indicador se desplace en el gel de resolución. Cuando el indicador alcance la parte inferior del gel, detener la electroforesis. Retirar el montaje del gel del aparato y separar las placas de vidrio. Retirar los espaciadores, cortar y tirar el gel de apilamiento y proceder inmediatamente a la tinción.
DETECCION DE PROTEINAS EN GELES
Todas las etapas de tinción de geles se realizan a temperatura ambiente con agitación suave (por ej., en una placa de movimiento giratorio) en un recipiente apropiado.
Tinción con Coomassie - Es el método de tinción de proteínas más empleado, con un nivel de detección del orden de 1 a 10 mg de proteína por banda.
Solución colorante de Coomassie - Disolver 1,25 g de Azul brillante de Coomassie R-250 en 1 litro de una mezcla de agua, metanol y ácido acético glacial (5:4:1).
Solución de decoloración - Emplear una mezcla de agua, metanol y ácido acético glacial (5:4:1).
Procedimiento - Sumergir el gel en un exceso de Solución colorante de Coomassie y dejar en contacto por lo menos durante 1 hora. Eliminar la Solución colorante de Coomassie. Decolorar el gel con un exceso de Solución de decoloración. Cambiar la Solución de decoloración varias veces hasta que las bandas de proteínas teñidas se distingan nítidamente sobre un fondo claro. Cuanto más se lave, menor será la cantidad de proteína que se pueda detectar. La decoloración se puede acelerar agregando en la Solución de decoloración algunos gramos de resina de intercambio aniónico.
[NOTA: las soluciones ácido-alcohólicas empleadas en este procedimiento no fijan por completo las proteínas en el gel. Esto puede conducir a la pérdida de algunas proteínas de bajo peso molecular durante el proceso de tinción y decoloración de geles finos. La fijación permanente se obtiene introduciendo el gel en una mezcla de agua, metanol y ácido tricloroacético (5:4:1) durante 1 hora antes de introducirlo en la Solución colorante de Coomassie.]
Tinción con plata - Es el método más sensible para proteínas teñidas en geles y permite detectar bandas que contengan 10 a 100 ng de proteína.
Reactivo de nitrato de plata - A una mezcla de 3 ml de amoníaco concentrado y 40 ml de hidróxido de sodio 1 M, agregar 8 ml de una solución al 20% de nitrato de plata, gota a gota, y con agitación. Diluir con agua a 200 ml.
Solución de fijación. - A 250 ml de metanol, agregar 0,27 ml de solución de formaldehído al 35% y diluir con agua a 500 ml.
Solución de desarrollo - Diluir 2,5 ml de una solución al 2% de ácido cítrico y 0,27 ml de solución de formaldehído al 35% con agua a 500 ml.
Solución de bloqueo - Emplear una solución al 10% v/v de ácido acético.
Procedimiento - Introducir el gel en exceso en Solución de fijación durante 1 hora. Eliminar la Solución de fijación, agregarla de nuevo e incubar otra vez durante por lo menos 1 hora o durante toda la noche, si es conveniente. Eliminar la Solución de fijación y lavar el gel con abundante agua durante 1 hora. Embeber el gel durante 15 minutos en una solución de glutaraldehído al 1% v/v. Lavar el gel dos veces durante 15 minutos con abundante agua. Embeber el gel en Reactivo de nitrato de plata recientemente preparado, durante 15 minutos, en la oscuridad. Lavar el gel tres veces durante 5 minutos con abundante agua, sumergirlo durante aproximadamente 1 minuto en Solución de desarrollo hasta que se obtenga una tinción satisfactoria. Detener el desarrollo por incubación en Solución de bloqueo durante 15 minutos y lavar el gel con agua.
DESECADO DE GELES DE POLIACRILAMIDA SDS TEÑIDOS
Los geles se someten a diferentes tratamientos, dependiendo del método empleado para teñirlos. Cuando se emplea Tinción con Coomassie, luego de la etapa de decoloración, colocar el gel en una solución al 10% de glicerol durante por lo menos 2 horas, siendo posible una incubación durante toda la noche. Cuando se emplea Tinción con plata, al finalizar el proceso de lavado, sumergir los geles en una solución al 2% de glicerol, durante 5 minutos.
Sumergir dos hojas de celulosa porosa en agua durante 5 a 10 minutos, colocar una de las hojas en el cuadro de secado, levantar el gel cuidadosamente y colocarlo sobre la hoja de celulosa. Eliminar las burbujas de aire que eventualmente puedan haber sido retenidas y algunos ml de agua a lo largo de los bordes del gel. Recubrir con la segunda hoja de papel y eliminar nuevamente las posibles burbujas de aire retenidas. Colocar en estufa para completar el secado o dejar secar a temperatura ambiente.
DETERMINACION DE PESO MOLECULAR
El peso molecular de las proteínas se determina mediante comparación de sus respectivas movilidades con las de varios marcadores de peso molecular conocido. Para la calibración de los geles se dispone de mezclas de proteínas de peso molecular conocido, que permiten obtener una tinción uniforme. Las soluciones madre concentradas de proteínas de peso molecular conocido preparadas en la solución reguladora de muestra se aplican en el mismo gel contiene la muestra de la proteína a analizar.
Inmediatamente después del desarrollo electroforético, señalar la posición del indicador, azul de bromofenol, para identificar el frente de migración de los iones. Después de la tinción, medir las distancias de migración de cada banda proteica (marcadores y muestras). Dividir la distancia de migración de cada proteína por la distancia de migración del indicador. Las distancias de migración normalizadas así obtenidas se denominan movilidades relativas de las proteínas (relativas al frente del indicador) y, por convención, se expresan como Rf . Graficar el logaritmo de los pesos moleculares relativos (M1) de las proteínas patrón en función de los valores de Rf. Los pesos moleculares desconocidos se pueden determinar por regresión lineal o por interpolación a partir de las curvas obtenidas; los valores obtenidos para las muestras se deben encontrar contenidos en la parte lineal del gráfico.
VALIDACION DEL ENSAYO
El ensayo sólo es válido si las proteínas empleadas como marcadores de pesos moleculares se distribuyen a lo largo del 80% de la longitud del gel y además si, en el intervalo de separación requerido, la separación obtenida para las bandas de proteínas relevantes, presenta una relación lineal entre el logaritmo de peso molecular y el Rf . En la monografía correspondiente se especifican los requisitos de validación adicionales referentes a la solución muestra.
CUANTIFICACION DE IMPUREZAS
Cuando en la monografía se especifica el límite de impurezas, se debe preparar una solución de referencia correspondiente al nivel de impureza especificado, por dilución de la solución muestra. En el electroforegrama obtenido a partir de la solución muestra, ninguna impureza (ni ninguna banda, a excepción de la principal) puede ser más intensa que la banda principal obtenida a partir de la solución de referencia.
Cuando se trabaja en las condiciones validadas, es posible cuantificar las impurezas por normalización con relación a la banda principal, empleando un densitómetro. En estos casos, se debe comprobar la linealidad de las repuestas.