630. METODOS DE FARMACOGNOSIA
Definición de Droga Vegetal - Se denomina así a las plantas o sus partes enteras, molidas o pulverizadas (flores, frutos, semillas, tubérculos, cortezas, etc.) frescas o secas, así como los jugos, gomas, látex, aceites esenciales o fijos y otros componentes similares, que se emplean puras o mezcladas en la elaboración de medicamentos.
Debido a las características de las drogas vegetales, en particular su falta de homogeneidad, se requieren procedimientos especiales en relación a los ensayos a realizar.
MUESTREO
Los siguientes procedimientos de muestreo constituyen las consideraciones mínimas aplicables a las drogas vegetales. Algunos productos o ensayos pueden requerir procedimientos más estrictos que incluyan el muestreo de mayor número de envases y/o más muestras por envase.
Si el examen externo de los envases y rótulos indica que puede considerarse el lote como homogéneo, tomar muestras individuales de un número de envases seleccionados aleatoriamente según se indica a continuación. Si el lote no puede considerarse homogéneo, fraccionarlo en sublotes que sean lo más homogéneos posible y realizar el muestreo con cada uno como un lote homogéneo.
|
N° de envases por lote (N) |
N° de envases a muestrear (n) |
|
1 a 5 6 a 50 > 50* |
todos 5 10 % de los envases |
* Redondear N al múltiplo de diez próximo superior.
Las muestras se deben tomar de las secciones superior, media e inferior de cada envase y en diferentes sitios. En el caso de los polvos o material compuesto por fragmentos de 1 cm o menos en cualquier dimensión, retirar la muestra a través de un dispositivo de muestreo que permita tomar el material desde la parte superior hasta el fondo del envase. Si el material está compuesto por fragmentos mayores de 1 cm en cualquier dimensión, retirar las muestras en forma manual. En el caso de fardos o bolsas grandes, las muestras deben tomarse a más de 10 cm de los bordes porque el contenido de humedad de la capa superficial puede ser diferente que el de las capas internas.
Combinar y mezclar las muestras tomadas de cada envase abierto, evitando aumentar el grado de fragmentación o modificar significativamente el contenido de humedad. Disponer la muestra así preparada en forma de cuadrado y fraccionarla diagonalmente en cuatro partes iguales. Tomar luego dos partes opuestas y mezclarlas cuidadosamente. Repetir el procedimiento, si fuera necesario, hasta obtener la cantidad requerida para realizar todos los ensayos necesarios (cuarteo).
Sólo si se indica, moler la muestra para que pase a través de un tamiz N° 20 y mezclar el polvo resultante. Si el material no puede ser molido, reducirlo al estado más fino posible.
ANALISIS MICROSCOPICO
Cortes y coloración
Hidratación o ablandamiento - Colocar en un vaso de precipitados una cantidad apropiada de material con 20 a 30 veces su volumen de agua. Colocar sobre una plancha calefactora o una tela metálica, calentar suavemente hasta ebullición y mantenerla durante 5 minutos. Si el material no puede ser cortado después de hidratarlo, se procede a ablandarlo hirviéndolo durante 5 minutos en agua con detergente y ensayando su consistencia. Si se considera que aún no está lo suficientemente blando como para ser cortado, colocar una cantidad apropiada del material en un vaso de precipitados que contenga un volumen apropiado de etilenglicol. Ensayar periódicamente la consistencia del material. Para futuros análisis, determinar el tiempo que tarda cada material en adquirir una consistencia tal que permita su corte.
Cortes - Obtener secciones transversales delgadas del material vegetal. Esto se logra cortando a mano alzada o mediante el empleo de micrótomo. [NOTA en el caso de la obtención de transcortes de hoja, resulta necesario el empleo de un soporte para poder cortar. Generalmente se coloca la hoja entre dos semicilindros de médula de sauco o de hinojo y se procede a cortar todo junto.]
Los instrumentos cortantes pueden ser hoja de afeitar, bisturí o cuchilla para histología.
Los cortes se colocan en un recipiente con agua (vidrios de reloj, vasos de precipitados de 30 ml). Se seleccionan los más delgados para observación al microscopio a 10x.
Coloración - Sumergir los cortes en solución de hipoclorito de sodio al 50 % para eliminar el contenido celular. Dejar actuar hasta que los cortes se vuelvan transparentes (no más de 10 a 15 minutos). Lavar los cortes con agua hasta eliminación del hipoclorito de sodio, pH neutro. Colocar los cortes en solución de azul de toluidina al 0,05 %, durante 10 segundos. Lavar con agua luego con solución de ácido acético al 0,5% y por último nuevamente con agua. Colocar entre porta y cubreobjetos con 2 a 3 gotas de una mezcla de glicerina-agua (1:1) y observar al microscopio a 10x y 40x. Las paredes celulósicas se tiñen de rosa púrpura. Las paredes lignificadas y las paredes con tanino se tiñen de color azul verdoso brillante. [NOTA: la coloración así obtenida no es estable.]
Observación de la droga en polvo
Observación directa - Colocar 2 a 10 mg de la droga en polvo sobre un portaobjetos. Agregar 2 ó 3 gotas de solución de ácido láctico al 5 % (diafanizante) y colocar el cubreobjetos. Observar al microscopio a 10x y 40x.
Reacciones histoquímicas -
Detección de almidón - Colocar 2 a 10 mg de la droga en polvo sobre un portaobjetos. Verter 2 ó 3 gotas de Solución de Lugol (SR) diluida (1:5) en agua. Colocar el cubreobjetos y observar al microscopio a 10x y 40x. Los granos de almidón se colorean de azulvioláceo intenso.
Detección de lípidos y aceites esenciales - Colocar 2 a 10 mg de la droga en polvo sobre un portaobjetos. Verter 2 ó 3 gotas de solución de Sudan III (SR) y dejar actuar durante 2 ó 3 minutos. Escurrir el líquido y lavar bien con alcohol 70 %. Colocar el cubreobjetos y observar al microscopio a 10x y 40x. Los lípidos aparecen como gotas de color rojo.
Detección de concreciones de carbonato de calcio (cistolitos) y de cristales de oxalato de calcio - Colocar 2 a 10 mg de la droga en polvo sobre un portaobjetos. Verter 2 ó 3 gotas de ácido clorhídrico 2 M, con la precaución de que el reactivo esté en íntimo contacto con todos los componente del polvo. Colocar el cubreobjetos y observar inmediatamente al microscopio a 10x. La presencia de carbonato de calcio está indicada por la aparición de burbujas. Los cristales de oxalato de calcio, que en general tardan más tiempo en disolverse, no desprenden burbujas.
Detección de taninos - Colocar 2 a 3 mg de la droga en polvo sobre un portaobjetos. Verter 2 ó 3 gotas de solución de cloruro férrico al 5 %. Colocar el cubreobjetos y observar al microscopio a 10x y 40x. La presencia de taninos se traduce en la aparición de masas oscuras de color pardo, azul o negro.
Obtención de disociados
Disociación. leve o débil - Este método se emplea principalmente para el análisis de hojas, tallos herbáceos y cortezas. Los cristales se conservan. Los almidones pierden su estructura característica.
Colocar en un vaso de precipitados de 30 ml una porción del material vegetal. Agregar 10 ml de solución de hidróxido de sodio al 5 % y llevar a ebullición durante 5 minutos. Enfriar. Trasvasar a un tubo de centrífuga. Centrifugar durante 2 minutos a 3000 rpm. Descartar la solución sobrenadante. Lavar con agua. Colocar una porción del centrifugado sobre un portaobjetos con 2 ó 3 gotas de una mezcla de glicerina-agua (1:1). Colocar el cubreobjetos y terminar la disgregación ejerciendo presión. Observar al microscopio a 10x y 40x.
Disociación fuerte - Este método se emplea principalmente para el análisis de leños, tegumentos de semillas y endocarpios esclerosados-carozos. No se conservan los cristales ni los almidones.
Colocar en un vaso de precipitados de 30 ml una porción del material vegetal. Agregar 10 ml de solución de hidróxido de potasio al 10 % y llevar a ebullición durante 10 minutos. Enfriar. Eliminar cuidadosamente el solución sobrenadante. Lavar con agua. Colocar el material vegetal en un tubo de centrífuga. Agregar 10 ml de solución de ácido crónico al 25 % y dejar actuar durante un tiempo no inferior a 30 minutos a temperatura ambiente. Ensayar la consistencia del material vegetal con una varilla de vidrio. Cuando esté lo suficientemente blando, centrifugar durante 2 minutos a 3000 rpm. Descartar el sobrenadante y lavar con agua hasta eliminar totalmente el color amarillo del ácido crómico. Colocar una porción del centrifugado sobre un portaobjetos con 2 ó 3 gotas de una mezcla de glicerina-agua (1:1). Colocar el cubreobjetos y terminar la disgregación ejerciendo presión. Observar al microscopio a 10x y 40x.
Determinación del índice de estomas
(Se emplea para hojas). Es el número de estomas por cada 100 células epidérmicas en un área fija.
Delimitar sobre una hoja de papel, con ayuda de una cámara clara, de un tubo de dibujo o de un ocular de dibujo, un área de 2 mm de lado empleando un micrómetro objetivo, observando con objetivo de 5x y ocular de 8x.
Colocar un trozo de hoja de 0,5 cm x 0,5 cm en un vaso de precipitados de 30 ml. Agregar 10 ml de una mezcla de hidrato de cloral y agua (5:2). Llevar a ebullición durante 10 a 15 minutos o hasta que el trozo se vuelva transparente. Esta operación se realiza bajo campana.
Colocar el trozo de hoja sobre un portaobjetos con la epidermis inferior hacia arriba. Agregar 2 ó 3 gotas de la mezcla de hidrato de cloral y agua y colocar el cubreobjetos. Contar las células epidérmicas y los estomas que aparecen en el área delimitada empleando un objetivo de 40x. Las dos células estomáticas se cuentan como una sola.
El índice de estomas se calcula por la fórmula siguiente:
|
S |
|
|
–––––––– |
100 |
|
S + E |
en la cual S es el número de estomas y E el número de células epidérmicas en el área delimitada.
METODOS DE ANALISIS
Materia extraña
Se considera materia extraña a cualquier parte de la planta medicinal que no esté comprendida en la definición o en la descripción de la monografía correspondiente; cualquier organismo, parte o producto de un organismo no comprendido en la definición o en la descripción; o residuos minerales, como por ej., tierra, piedras, arena o polvo.
Durante el almacenamiento, los productos deben mantenerse en un área limpia, de modo de evitar su contaminación. Deben tomarse precauciones especiales para evitar la proliferación de hongos dado que algunos de ellos pueden generar aflatoxinas.
A menos que se especifique de otro modo en la monografía correspondiente, obtener por cuarteo las siguientes cantidades de muestra:
|
Raíces, rizomas, cortezas y plantas enteras |
500 g |
|
Hojas, flores, semillas y frutos |
250 g |
|
Drogas vegetales en fragmentos de 0,5 g o menores |
50 g |
Extender la muestra en una capa delgada y separar la materia extraña a mano, en la forma más completa posible. Pesarla y determinar el porcentaje de materia extraña a partir de la cantidad de droga empleada.
Cenizas totales
Pesar exactamente una cantidad de muestra, obtenida según se indica en Muestreo, que represente de 2 a 4 g del material; molerla para que pase a través de un tamiz N° 20 y secarla al aire en un crisol previamente pesado. Someter a calcinación, suavemente al principio, y aumentar gradualmente la temperatura hasta 675 x 25 °C. Continuar la calcinación hasta eliminar él residuo carbonoso y determinar el peso de las cenizas. Si de esta forma no se obtienen cenizas libres de residuo carbonoso, extraer la masa carbonizada con agua caliente. Recolectar el residuo insoluble en un papel de filtro libre de cenizas, calcinar el residuo y el papel de filtro hasta que las cenizas sean blancas o casi blancas. Luego, agregar el filtrado, evaporarlo hasta sequedad y calentar a 675 ± 25 °C. Si de esta forma no se obtienen cenizas libres de residuo carbonoso, enfriar el crisol, agregar 15 ml de alcohol, disgregar las cenizas con una varilla de vidrio, quemar el alcohol y calentar nuevamente a 675 ± 25 °C. Enfriar en un desecador, pesar las cenizas y calcular el porcentaje de cenizas totales a partir de la cantidad de droga empleada.
Cenizas insolubles en ácido
Calentar a ebullición las cenizas obtenidas según se indica en Cenizas totales, con 25 ml de ácido clorhídrico 3 M durante 5 minutos. Recolectar el material insoluble en un crisol filtrante previamente pesado o en un filtro libre de cenizas lavado con agua caliente, llevar a temperatura de calcinación y pesar. Determinar el porcentaje de cenizas insolubles en ácido a partir del peso de droga empleada.
Extracto alcohólico
Método I (método de extracción caliente) - Transferir a un erlenmeyer con tapón de vidrio aproximadamente 4 g, exactamente pesados, del material en polvo grueso y secado al aire. Agregar 100 ml de alcohol y pesar el erlenmeyer. Agitar y dejar en reposo durante 1 hora. Conectar un refrigerante al erlenmeyer y calentar suavemente a ebullición durante 1 hora, enfriar y pesar. Ajustar nuevamente al peso original mediante el agregado de alcohol. Agitar y filtrar rápidamente a través de un filtro seco. Transferir 25 ml del filtrado a un cristalizador y evaporar hasta sequedad en baño de agua. Secar a 105 °C durante 6 horas, enfriar en un desecador durante 30 minutos y pesar inmediatamente. Calcular el contenido, en mg por g, de materia extraible en alcohol en la muestra empleada.
Método II (método de extracción fría) - Transferir a un erlenmeyer con tapón de vidrio aproximadamente 4 g, exactamente pesados, de material en polvo grueso y secado al aire. Agregar 100 ml de alcohol, tapar el erlenmeyer y macerar durante 24 horas, agitando frecuentemente durante las primeras 8 horas y luego dejar reposar. Filtrar rápidamente, tomando precauciones para evitar la pérdida de alcohol. Evaporar 25 ml del filtrado hasta sequedad en un cristalizador previamente pesado y secar a 105 °C hasta peso constante. Calcular el contenido, en mg por g, de la materia extraible en alcohol en la muestra empleada.
Extracto acuoso
Método I (método de extracción caliente) - Proceder según se indica para el Método I en Extracto alcohólico, excepto que se debe emplear agua en lugar de alcohol.
Método II (método de extracción fría) - Proceder según se indica para el Método II en Extracto alcohólico, excepto que se debe emplear agua en lugar de alcohol.
Fibra cruda
Extraer con éter hasta agotar una cantidad exactamente pesada de la muestra que represente aproximadamente 2 g de la droga. Transferir la droga agotada a un balón de 500 ml y agregar 200 ml de ácido sulfúrico diluido (1:78) en agua a punto de ebullición. Conectar el balón a un refrigerante y calentar a reflujo la mezcla durante exactamente 30 minutos. Filtrar a través de un filtro de papel grueso o muselina y lavar el residuo retenido en el filtro con agua hirviendo hasta que los lavados no sean ácidos. Lavar el residuo en el balón con 200 ml de solución de hidróxido de sodio, libre de carbonato de sodio, aproximadamente a 100 °C, ajustada al 1,25 % mediante titulación. Calentar nuevamente a reflujo la mezcla durante exactamente 30 minutos, filtrar rápidamente a través de un filtro previamente pesado, lavar el residuo con agua hirviendo hasta que el último lavado sea neutro y secar a 110 °C hasta peso constante. Someter el residuo seco a calcinación hasta peso constante, enfriar en un desecador y pesar las cenizas obtenidas: la diferencia entre el peso obtenido por secado a 110 °C y el de las cenizas representa el peso de la fibra cruda. [NOTA: la ebullición con ácido y álcali debería continuar durante exactamente 30 minutos, desde el tiempo que el liquido (que se enfría debajo del punto de ebullición al agregarlo al balón frío) hierve nuevamente. Luego de que la solución haya entrado en ebullición, se debe disminuir el calor lo suficiente como para que ésta se mantenga.
Durante la ebullición, rotar el balón suavemente, varias veces, para remover cualquier partícula que pueda quedar adherida a las paredes. Una corriente lenta de aire introducida en el balón durante la operación ayuda a impedir la excesiva formación de espuma].
Determinación de aceites esenciales
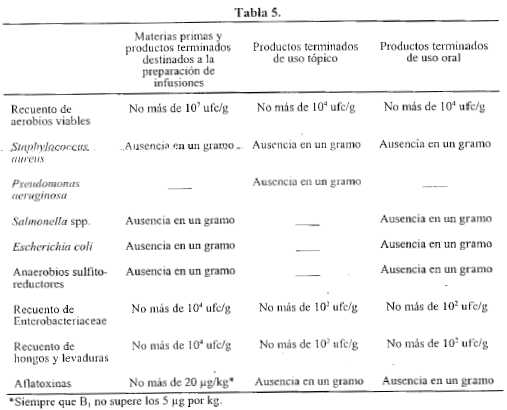
La determinación de aceites esenciales en vegetales se lleva a cabo mediante extracción por arrastre con vapor de agua en un aparato apropiado en las condiciones que se detallan a continuación. El aceite esencial es recolectado en un tubo graduado empleando xileno para fijarlo, mientras que el agua retorna al balón de extracción.
Aparato (ver Figura I) - Consta de un balón con cuello corto esmerilado cuyo diámetro máximo interno es de 29 mm y de un condensado con junta esmerilada. Las diferentes partes de este condensador están construidas en una sola pieza de vidrio de bajo coeficiente de expansión. El tapón K’ tiene una abertura y el tubo K posee un orificio de 1 mm de diámetro, el cual coincide con la ubicación de la abertura del tapón. El extremo final del tubo K es esmerilado y tiene un diámetro interno de 10 mm; un tubo en forma de pera J de 3 ml de capacidad; un tubo JL graduado en 0,01 ml; un tubo en forma de bulbo de aproximadamente 2 ml de capacidad y una válvula de tres vías M. La unión B se encuentra a 20 mm de la graduación máxima superior. El aparato posee un dispositivo apropiado para ser calentado.
Procedimiento - Llevar a cabo el ensayo de acuerdo con la naturaleza de la droga a ser examinada. Transferir el balón el volumen de líquido indicado en la monografía correspondiente, agregar algunos trozos de plato poroso y colocar el condensador. Introducir agua a través del tubo de llenado N hasta el nivel B. Quitar el tapón K’ y transferir la cantidad indicada de xileno empleando una pipeta con su extremo en la parte inferior del tubo K. Colocar el tapón K’ asegurándose que los orificios de K y K’ coincidan entre sí. Calentar el líquido en el balón hasta ebullición y ajustar la velocidad de extracción a aproximadamente 2 ml por minuto, a menos que se especifique de otro modo en la monografía correspondiente.
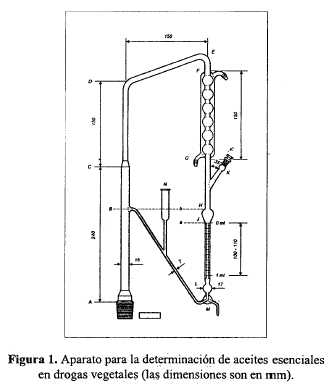
Para determinar la velocidad de extracción, disminuir el nivel de agua por medio de la válvula de tres vías hasta que el menisco se encuentre en la marca inferior a (ver Figura 2). Cerrar la válvula y medir el tiempo que toma el líquido en alcanzar la marca superior b. Abrir la válvula y continuar con la extracción, modificando el calentamiento para regular la velocidad de extracción: Extraer durante 30 minutos. Detener el calentamiento y leer el volumen de xileno en el tubo graduado después de por lo menos 10 minutos.
Transferir el balón la cantidad de muestra especificada en la monografía correspondiente y continuar la extracción como se ha descripto anteriormente en tiempo y velocidad según se indique. Detener el calentamiento y después de 10 minutos leer el volumen de líquido recolectado en el tubo graduado y restarle el volumen de xileno anteriormente medido. Calcular el resultado en ml por cada 100 g de droga.
Cuando un aceite esencial se emplee para propósitos analíticos, la obtención de la mezcla de xileno y aceite esencial libre de agua se realiza como se detalla a continuación: quitar el tapón K’ y transferir 1,1 ml de una solución de fluoresceinato de sodio al 0,1% y 0,5 ml de agua. Disminuir el volumen de la mezcla de xileno y aceite esencial dentro del tubo L por medio de la válvula de tres vías; dejar en reposo durante 5 minutos y descargar la mezcla lentamente hasta alcanzar justo el nivel de la válvula M. Abrir la válvula en el sentido contrario a las agujas del reloj de manera tal que el agua fluya fuera del tubo de conexión BM. Lavar el tubo, primero con acetona y luego con tolueno, introducidos por el tubo de llenado N. Girar la llave en el sentido contrario a las agujas del reloj de manera tal que se pueda recuperar la mezcla de xileno y aceite esencial en un recipiente apropiado.
Pérdida por secado
Reducir 10 g de muestra a fragmentos de aproximadamente 3 mm de espesor. Las semillas o frutos más pequeños de 3 mm se deben fragmentar. Evitar el empleo de molinos de gran velocidad para preparar la muestra y tomar las precauciones necesarias para no modificar el contenido de humedad de la muestra. Pesar exactamente 10 g de droga en un cristalizador previamente pesado. Secar a 105 °C durante 5 horas y pesar. Repetir el procedimiento de secado y pesado a intervalos de 1 hora hasta que la diferencia entre dos pesadas sucesivas corresponda a no más de 0,25 % de muestra.
RESIDUOS DE PESTICIDAS
La lista de pesticidas consignada para este ensayo es de carácter orientativo. Para mayor información consultar las resoluciones que a tal efecto emita la Secretaría de Agricultura, Ganadería, Pesca y Alimentos de la Nación.
Definición - Para los propósitos de esta Farmacopea, un pesticida es aquella sustancia o mezcla de sustancias empleadas para prevenir, destruir o controlar cualquier plaga, especies de plantas o animales indeseables que puedan causar daño o interferir en la producción, procesamiento, almacenamiento, transporte o comercialización de las drogas vegetales. El término incluye a sustancias empleadas como reguladores del crecimiento, desfoliantes o desecantes y cualquier otra sustancia aplicada para brindar una protección antes o después de la cosecha, o para prevenir el deterioro de la droga vegetal durante el almacenamiento o transporte.
Límites - A menos que se especifique de otro modo en la monografía correspondiente la muestra debe cumplir con los límites expresados en la Tabla 1. Los pesticidas que no figuren en la misma deben cumplir con las especificaciones dadas por las resoluciones que a tal efecto emita la Secretaría de Agricultura, Ganadería, Pesca y Alimentos de la Nación. Los límites para los pesticidas que no figuran en la Tabla 1 se calculan por la fórmula siguiente:
|
IDA x M |
|
––––––––––––– |
|
DDD x 100 |
en la cual IDA es la ingesta diaria admisible, recomendada por la FAO, expresada en mg por kg de peso corporal, M es el peso corporal en kilogramo (considerar 60 kg) y DDD es la dosis diaria de la droga en kg.
Si la droga vegetal es empleada en la preparación de extractos, tinturas u otras formas farmacéuticas cuyo método de preparación modifique el contenido del pesticida en el producto final, el límite se calcula por la fórmula siguiente:
|
IDA x M x E |
|
––––––––––––– |
|
DDD x 100 |
en la cual E es el factor de extracción del método de preparación, determinado experimentalmente.

Análisis cualitativo y cuantitativo de residuos de pesticidas
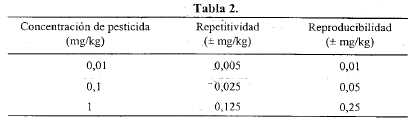
Los procedimientos analíticos empleados deben satisfacer los siguientes criterios: el método de extracción elegido debe ser apropiado para la combinación de pesticidas que se pretende investigar y no provocar interferencias. Los límites de detección y cuantificación deben determinarse para cada combinación de pesticidas a ser analizada. La recuperación debe estar entre el 70 y 110 %. La repetitividad y reproducibilidad del método no debe ser menor que la indicada en la Tabla 2.
Pesticidas organoclorados, organofosforados y piretroides
Los ensayos que se describen a continuación se emplean para el análisis de pesticidas, a menos que se especifique lo contrario en la monografía correspondiente. Dependiendo de la sustancia a analizar, puede ser necesario, en algunos casos, introducir modificaciones en los procedimientos descritos. En cualquier caso, puede ser necesario emplear otra columna con diferente polaridad u otro método de detección (espectrometría de masa, etc.) o un método diferente para confirmar los resultados obtenidos (métodos inmunoquímicos, etc.).
Este ensayo es válido solamente para el análisis de drogas vegetales con un contenido de agua menor de 15%. Las muestras con mayor humedad pueden secarse, teniendo en cuenta que el procedimiento empleado no afecte significativamente el contenido de pesticida.
Extracción - A 10 g de muestra, en forma de polvo grueso, agregar 100 ml de acetona y dejar reposar durante 20 minutos. Agregar 1 ml de solución que contenga 1,8 µg por 1 ml de carbofenotion en tolueno. Homogeneizar empleando un agitador de alta velocidad durante 3 minutos. Filtrar y lavar el residuo con dos porciones de acetona de 25 ml. Combinar el filtrado y los lavados en un balón y evaporar hasta casi sequedad en un evaporador rotatorio a una temperatura no mayor a 40 °C. Al residuo así obtenido, agregarle unos ml de tolueno y continuar con el calentamiento a la temperatura especificada anteriormente hasta evaporación total de la acetona. Disolver el residuo en 8ml de tolueno. Filtrar a través de una membrana filtrante de 45 µm, lavar el balón y el filtrado de tolueno. Diluir a 10 ml con el mismo solvente. Denoninar esta solución como Solución A.
Purificación - Pesticidas organoclorados, o ganofosforados y piretroides - Emplear una columna de 30 cm x 7,8 mm para cromatografía (ver 100. Cromatografía) con fase estacionaria constituida por copolímero rígido, esférico de divinilbenceno y estireno, de 5 µm de diámetro. Emplear tolueno como fase móvil. El caudal es de aproximadamente 1 ml por minuto.
Para verificar la aptitud de la columna, inyectar 100 µl de una solución en tolueno que contenga 0,5 mg por ml de rojo de metilo y 0,5 mg por ml de azul de oracet 2R y proceder con la cromatografía. La columna es apta si los colores del eluato cambian de anaranjado a azul en un volumen de elución de aproximadamente 10,3 ml. Calibrar la columna, si fuera necesario, empleando una solución en tolueno que contenga una concentración apropiada del pesticida a ser analizado de menor peso molecular (por ej., diclorvos) y aquél de mayor peso molecular (por ej., deltametrina). Determinar en qué fracción del eluato se encuentran ambos pesticidas.
Purificación de la solución - Inyectar un volumen apropiado de Solución A (100 a 500 µl) y proceder con la cromatografía. Recolectar las fracciones según se indicó anteriormente e identificarlas como Solución B. Los pesticidas organofosforados generalmente eluyen entre 8,8 y 10,9 ml, y los organoclorados y piretroides lo hacen entre 8,5 y 10,3 ml.
Pesticidas organoclorados y piretroides - Emplear una columna cromatográfica de 10 cm x 5 mm. Introducir un trozo de lana de vidrio y 0,5 g de gel de sílice para cromatografia tratada según se indica a continuación: calentar el gel de sílice para cromatografia en una estufa a 150 °C durante 4 horas. Dejar enfriar y agregar gota a gota una cantidad de agua equivalente a 1,5 de la masa de gel de sílice empleada, agitar vigorosamente hasta que desaparezcan los grumos y continuar agitando durante 2 horas empleando un agitador mecánico. Acondicionar la columna empleado 1,5 ml de hexano. [NOTA: pueden emplearse columnas empacadas con 0,5 g de gel de sílice apropiado si su empleo ha sido previamente validado].
Concentrar la Solución B hasta casi sequedad bajo una corriente de helio o nitrógeno libre de oxígeno y diluir a un volumen apropiado con tolueno (200 µl a 1 ml de acuerdo con el volumen inyectado en la preparación de la Solución B). Transferir cuantitativamente a la columna y eluir con 1,8 ml de tolueno. Recolectar el eluato e indentificarlo como Solución C.
Análisis cuantitativo
Pesticidas organofosforados -
Sistema cromatográfico - Emplear un equipo para cromatografía de gases con un detector de nitrógeno-fósforo o un detector de ionización a la llama y una columna de sílice fundida de 30 m x 0,32 mm con una fase estacionaria constituida por una capa de dimetilpolisiloxano de 0,25 µm de espesor. Mantener la columna a 80 °C durante 1 minuto, luego aumentar la temperatura a razón de 30 °C por minuto hasta 150°C, mantener a esa temperatura durante 3 minutos. Aumentar la temperatura a razón de 4°C por minuto hasta 280 °C y mantener a esta temperatura durante 1 minuto. Mantener el inyector y el detector a 250 y 275 °C, respectivamente. Se emplea hidrógeno como gas transportador.
[NOTA 1: pueden emplearse otros gases transportadores como nitrógeno o helio si su empleo ha sido previamente validado].
[NOTA 2: emplear carbofenotion como estándar interno; en ciertos casos puede ser necesario emplear un segundo estándar interno para identificar una posible interferencia con el pico correspondiente al carbofenotion].
Soluciones estándar - Preparar al menos tres soluciones en tolueno, que contengan los insecticidas a determinar y carbofenotion en concentraciones apropiadas para obtener una curva de calibración.
Solución muestra - Concentrar la Solución B bajo una corriente de helio o nitrógeno libre de oxígeno hasta casi sequedad y diluir a 100 µl con tolueno.
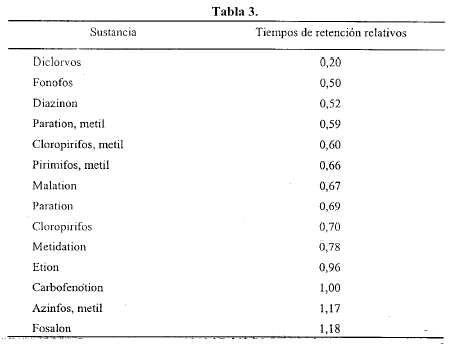
Procedimiento - Inyectar por separado en el cromatógrafo los volúmenes elegidos para cada solución, registrar los cromatogramas y medir las respuestas de los picos. Los tiempos de retención relativos aproximados se indican en la Tabla 3. [NOTA: los valores tabulados son orientativos y se transcriben para dar una idea de la secuencia en que eluyen las sustancias].
Calcular el contenido de pesticida a partir de las respuestas de los picos y las concentraciones de las soluciones empleadas.
Pesticidas organoclorados y piretroides - Sistema cromatográfico - Emplear un cromatógrafo de gases equipado con un detector de captura electrónica y una columna de sílice fundida de 30 m x 0,32 mm con fase estacionaria constituida por una capa de dimetilfenilpolisiloxano de 0,25 µm de espesor. Mantener la columna a 80 °C durante 1 minuto, aumentar la temperatura a razón de 30 °C por minuto hasta 150 °C, mantener esa temperatura durante 3 minutos. Aumentar la temperatura a razón de 4 °C por minuto hasta 280 °C y mantener a esta temperatura durante de 1 minuto. Mantener el inyector y el detector a 250 y 275 °C, respectivamente. Se emplea hidrógeno como gas transportador.
[NOTA 1: pueden emplearse otros gases transportadores como nitrógeno o helio si su empleo ha sido previamente validado].
[NOTA 2: emplear carbofenotion como estándar interno, en ciertos casos puede ser necesario emplear un segundo estándar interno para identificar una posible interferencia con el pico correspondiente al carbofenotion).
Soluciones estándar - Preparar al menos tres soluciones en tolueno, que contengan los pesticidas a determinar y carbofenotion en concentraciones apropiadas para obtener una curva de calibración.
Solución muestra - Concentrar la Solución C bajo una corriente de helio o nitrógeno libre de oxígeno hasta casi sequedad y diluir a 500 µl con tolueno.
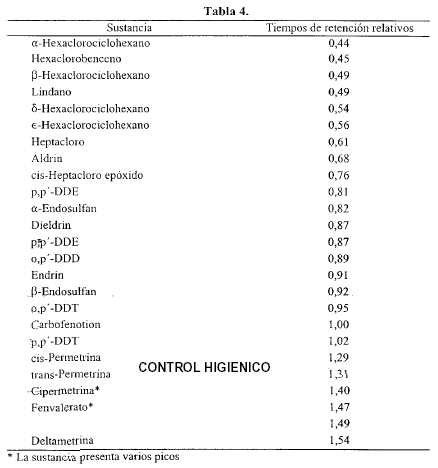
Procedimiento - Inyectar por separado en el cromatógrafo los volúmenes elegidos para cada solución, registrar los cromatogramas y medir las respuestas de los picos. Los tiempos de retención relativos aproximados se indican en la Tabla 4. [NOTA: los valores tabulados son orientativos y se transcriben para dar una idea de la secuencia en que eluyen-las sustancias].
Calcular el contenido de pesticida a partir de las respuestas de los picos y las concentraciones de las soluciones empleadas.
Proceder según se indica en <90>. Control higiénico de productos no obligatoriamente estériles y en <110>. Determinación de aflatoxinas. Los límites permitidos son los establecidos en la Tabla 5.