
20. ANALISIS TERMICO
Este análisis permite obtener información sobre propiedades y transformaciones físicas y/o químicas de una muestra cuando es sometida a variaciones de temperatura en una atmósfera específica, como ser: características de los cristales, estado, transformaciones polimórficas, transiciones vítreas, temperaturas y calores específicos de transición y de fusión, fenómenos de sublimación, interacciones sólido-sólido, etc. La medición instrumental de estos fenómenos tiene la ventaja de poseer alta sensibilidad, precisión y exactitud.
El análisis térmico permite la identificación, control de pureza y estabilidad de las sustancias, ya que las transiciones de estado ocurren a temperaturas características para cada una de ellas. Las técnicas de Análisis Térmico que se emplean con mayor frecuencia son: la Calorimetría Diferencial de Barrido (CDB), el Análisis Térmico Diferencial (ATD), el Análisis Termogravimétrico (ATG) y el Análisis Termomecánico (ATM). Las propiedades medidas por estas técnicas se indican en la Tabla 1.

La información dada por las diferentes técnicas se resume en la Tabla 2, excepto para el Análisis Térmico Diferencial, cuyas aplicaciones varían según las características de los aparatos.

Es necesario incluir en cada registro térmico una descripción completa de las condiciones empleadas, entre las que se encuentran: marca y modelo del aparato, registro de la última calibración, tamaño e identificación de la muestra, material, capacidad y estado del crisol empleado para contener la muestra, composición y caudal del gas empleado, presión del sistema, programa de temperaturas y sensibilidad de las determinaciones.
Calorimetría diferencial de Barrido (CDB)
Este análisis mide la absorción o desprendimiento de calor producida durante el calentamiento o enfriamiento de una muestra (procesos dinámicos), o durante el mantenimiento de la misma a una temperatura fija (proceso isotérmico), detectando cualquier fenómeno (transiciones físicas o reacciones químicas) acompañado por una entalpía. El calentamiento se produce en un Horno provisto de un sensor altamente sensible, que permite medir la diferencia entre los flujos de calor de la muestra y un crisol de referencia.
Análisis de impurezas eutécticas -
La fusión de un compuesto cristalino puro debe producirse dentro de un intervalo de temperatura muy reducido. La presencia de impurezas eutécticas produce una expansión del intervalo de fusión de las mismas. Este fenómeno puede verificarse a través de los registros térmicos de muestras con diferentes porcentajes de impurezas, en los cuales el intervalo de fusión se amplía a medida que aumenta la concentración de éstas, disminuyendo a su vez la temperatura del pico de fusión. Un material con 99% de pureza funde aproximadamente en un 20% a una temperatura 3°C por debajo del punto de fusión del material puro. El fundamento de la determinación de pureza de una sustancia se basa en el mencionado fenómeno, que relaciona la disminución del punto de fusión con la cantidad de impurezas presentes en la misma.
Las características de las impurezas eutécticas es que son solubles en la fase líquida formada durante la fusión, pero no lo son en la fase sólida del componente principal. Son necesarias semejanzas químicas para que se produzca la solubilidad en el material fundido. Las impurezas de síntesis generalmente son similares al producto final y no presentan problemas de solubilidad en el material fundido.
La evaluación de la curva de fusión por Calorimetría Diferencial de Barrido, a través de la ley de Van’t Hoff sobre la depresión del punto de fusión de sistemas eutécticos, en su forma simplificada [ver ecuación (1)], permite obtener los parámetros de fusión (temperatura, intervalo y calor de fusión, y cálculo de la pureza eutéctica) con muestras del orden del miligramo. La simplificación es apropiada, considerando que la expansión del intervalo de fusión es pequeña cuando la concentración de impurezas es baja.
Se obtiene así la siguiente relación entre la fracción molar de la impureza y la disminución del punto de fusión:
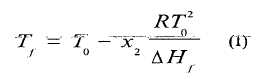
en la cual Tf la temperatura de la muestra en el proceso de fusión, en K, durante el equilibrio entre los cristales sólidos del componente principal y la fase líquida, T0 es la temperatura de fusión, en K, del componente principal puro, x2 es la fracción molar del componente minoritario (impureza) en la fase líquida, durante el proceso de fusión, DH es el calor molar de fusión del componente principal y R es la constante de los gases (R = 8,134 J/K mol)f.
Analizando el proceso de calentamiento de una muestra con impurezas eutécticas, a través de un diagrama de fases, se observa que el total de la impureza funde a la temperatura eutéctica, por arriba de la cual la fase sólida consiste sólo en el componente principal puro. En tanto la temperatura se aproxima al punto de fusión Tf, la fracción molar de la impureza en la fase líquida x2 disminuye constantemente ya que el componente principal puro se disuelve en la solución eutéctica durante este intervalo, verificándose la ecuación (2):
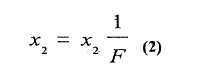
en la cual x2 es la fracción molar de la impureza en la fase líquida en cualquier punto de equilibrio durante el proceso de fusión, x2 * es la fracción molar de la impureza en la fase líquida de la sustancia completamente fundida o en la sustancia original y F es la fracción fundida.
En la ecuación (2) se observa que la concentración de la impureza en la fase líquida, a cualquier temperatura de equilibrio durante la fusión, es inversamente proporcional a la fracción fundida a esa temperatura.
En la temperatura de fusión, Tf, F es igual a 1, y x2 es iguala x2.*.
La combinación de las ecuaciones (1) y (2) da origen a la ecuación (3):
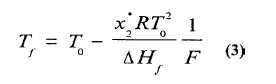
Según esta función, el gráfico de las temperaturas de equilibrio Tf de la muestra durante la fusión, en función de la inversa de la fracción fundida 1/F, es una línea recta cuya pendiente es igual a la disminución del punto de fusión. El punto de fusión teórico de la sustancia pura se obtiene mediante la extrapolación a 1/F igual a cero. Las desviaciones de la linealidad de esta recta son corregidas a través de factores que modifican los valores de DH f obtenidos.
En esta expresión se observa que la disminución del punto de fusión es directamente proporcional a la fracción molar de la impureza.
Estas evaluaciones se aplican con exactitud cuando las impurezas no exceden el 2% en moles.
Las impurezas no eutécticas no son evaluables a través de CDB. Consisten en moléculas que presentan la misma forma, tamaño y carácter que el componente principal y se acomodan en la matriz del cristal del componente principal sin modificación de su estructura, situación. favorecida en cristales menos ordenados, cuyos valores de fusión son más bajos. En tales casos, las estimaciones de pureza arrojan valores más altos que los reales. Su efecto puede ser incluso el de aumentar el punto de fusión. Ejemplos de impurezas no eutécticas son los cristales mixtos y las soluciones sólidas.
A las sustancias que presentan estados polimórficos no se les determina la pureza, a menos que se conviertan completamente en una de las modificaciones estables durante la fusión.
Por otro lado, la CDB y el ATD son intrínsecamente útiles para detectar y controlar el polimorfismo.
El análisis de pureza no debe aplicarse a muestras que funden presentando simultáneamente fenómenos de evaporación y/o descomposición.
Análisis Térmico Diferencial (ATD)
Este análisis mide la diferencia de temperatura entre la muestra en ensayo y una referencia inerte, ambas calentadas bajo las mismas condiciones, mientras que la CDB permite cuantificar las absorciones y desprendimiento de calor. Actualmente, de los análisis de ATD también pueden obtenerse resultados calorimétricos cuantificables.
Análisis termogravimétrico (ATG)
Este análisis registra el peso de la muestra en función de la temperatura o del tiempo de calentamiento, mediante el empleo de una termobalanza. Incluye programas de calentamiento dinámico o de temperatura fija (proceso isotérmico). Suministra más información que la pérdida por secado a una temperatura determinada, ya que detecta las temperaturas a las que se desprenden las sustancias volátiles retenidas, además de cuantificar los respectivos desprendimientos.
Muchas sustancias tienen la capacidad de formar hidratos y/o solvatos. En los primeros, el agua está presente no sólo en su superficie como humedad, sino también en el cristal. Esta propiedad, conocida como pseudopolimorfismo, puede conducir a complejos procesos de fusión.
Generalmente, la pérdida de solvente adsorbido en la superficie puede distinguirse de la pérdida de solvente ocluido en el cristal y de las pérdidas de masa producidas por descomposición de la sustancia.
Las mediciones se llevan a cabo bajo reflujo programado de un gas apropiado. El contenido porcentual de pérdida, G, se calcula por la fórmula siguiente:
G (% de pérdida) = 100 Dm/m0
en la cual Dm es la pérdida de masa y m0 es el peso inicial de la muestra.
Dado que el Análisis Termogravimétrico no identifica específicamente los productos de reacción, pueden analizarse los gases desprendidos con metodologías apropiadas. También se emplea para la caracterización de sustancias la combinación de CDB y ATG.
Aparato - Consta de una microbalanza asociada a una fuente de calor programable. Los aparatos difieren, principalmente, en el intervalo de masas aceptable para las muestras a analizar y la forma de detección de la temperatura de la muestra. Deben realizarse calibraciones periódicas de las determinaciones de masa, mediante el empleo de pesas patrón y de la escala de temperatura, empleando Sustancias de referencia apropiadas.
Análisis Termomecánico (ATM)
Este análisis mide los cambios dimensionales de una muestra bajo la acción de pequeñas cargas (modo dilatométrico), en función de la temperatura o el tiempo. Además de la detección de las transiciones vítreas, es importante el cálculo de los parámetros Coeficiente local de expansión, Conversión y Coeficiente medio de expansión.