
CAPITULO 8
PATOLOGIAS HABITUALES
1. REHOSPITALIZACION
Diana Rodríguez
Es conocido que los recién nacidos de alto riesgo, en especial los prematuros menores de 1500 gramos al nacer, están expuestos a una tasa 2 a 5 veces más alta de rehospitalizaciones que los de mayor peso durante el primer año de vida, presentando, aproximadamente un 40%, por lo menos una internación en ese período, valor que se mantiene más alto que la población general hasta los 5 años de edad.
Probablemente este problema se incremente en la medida que aumente la sobrevida en los prematuros de bajo peso extremo (menos de 1000g).
La incidencia de reinternación en nuestro medio, al igual que en otros países, varía sensiblemente de un centro a otro, dependiendo, principalmente, de las características de la población asistida.
Es muy importante que antes del alta hospitalaria, las familias de los pacientes con probabilidades de reinternación, sean prevenidas de que esto puede suceder, y a la vez aconsejadas respecto a distintas medidas que convienen tomar, tales como evitar la exposición al tabaco, no estar con personas que estén cursando enfermedades virales agudas, no asistir a lugares muy concurridos, desalentar la concurrencia a guarderías, sugerir la vacunación antigripal en los niños mayores de 6 meses y/ o en los convivientes que pudieran recibirla (de acuerdo a la época del año) y, muy especialmente, estimular la alimentación con leche materna. Hay evidencias de que la leche materna es uno de los principales factores protectores en este grupo de niños.
Algunos prematuros podrían recibir anticuerpos monoclonales humanizados para la prevención del virus sincicial respiratorio. (ver Capítulo Nº 3). Este factor, junto con el escaso peso y los bajos niveles de Ig G transferidos por la madre, son importantes aspectos que inciden en la reinternación de los prematuros pequeños.
Algunos factores de riesgo socioambientales, como el mal control prenatal, la falta de cobertura médica, la ausencia del padre en la casa, la presencia de hermanos conviviendo, la utilización de braseros dentro de la casa y la baja instrucción materna, podrían tener un importante rol en los índices de reinternación. Respecto a este último punto, se vio que en hijos de madres de buen nivel educativo pero adictas y/o solteras aumenta el riesgo.
La causa más frecuente de reinternación en prematuros en el primer año de vida es la respiratoria, y dentro de ella la bronquiolitis (la etiología preponderante es el virus sincicial respiratorio).
De acuerdo con algunas publicaciones, entre un 22% y 56% de los niños con displasia broncopulmonar, van a necesitar reinternación durante el primer año de vida por enfermedad aguda respiratoria, generalmente de origen viral y entre un 37% a 69% en el segundo año . Sin embargo es de hacer notar que la alta incidencia de reinternación por enfermedad respiratoria aguda, no queda limitada a los niños con DBP, ya que alrededor de un 25% de niños sin ella igualmente se reinternarán.
Las causas quirúrgicas ocupan un lugar importante como motivo de rehospitalización. La hernia inguinal (HI) predomina en prematuros, especialmente en varones, en una relación de 3/1 respecto a los de término, es bilateral en una relación 2/1 y es menos frecuente su atascamiento en una relación 1/2.
La HI derecha es más frecuente ya que el testículo de ese lado baja posteriormente. Junto con el hidrocele son patologías secundarias a la persistencia de un proceso vaginal permeable. Este es una proyección digitiforme del peritoneo que acompaña al testículo a medida que desciende hacia el escroto.
En la mujer, la extensión peritoneal acompaña al ligamento redondo y puede permanecer permeable, transformándose en saco herniario potencial. Con frecuencia se diagnostica por la observación de una masa ovoidea en la ingle. Esta representa el ovario herniado en el saco abierto. Aunque la gónada puede ser reducida nuevamente al abdomen, con frecuencia se prolapsa repetidamente hasta su corrección quirúrgica.
El hidrocele con frecuencia se asocia con HI, pero puede presentarse como un hallazgo aislado. El líquido puede estar en comunicación con la cavidad peritoneal y por lo tanto aumentar o disminuir de tamaño o estar separado y completamente aislado en el escroto.
En el caso de existir una HI con un anillo estrecho, el intestino puede quedar atascado, por lo que la cirugía debe programarse para el momento más temprano posible teniendo en cuenta las condiciones clínicas del paciente.
En alrededor del 60% de los pacientes es posible encontrar la hernia del lado opuesto o asintomática.
No se ha probado si un Programa de seguimiento es efectivo para disminuir la reinternación; pareciera ser que es el seguimiento personalizado de cada uno de los egresados de la terapia intensiva neonatal lo que ayudaría a disminuir el riesgo.
BIBLIOGRAFIA
Avery G. B. Neonatología. Fisiopatología y manejo del RN. 3ª Ed. Buenos Aires, Panamericana, 1990:977.
Cunningham C., Gross S. Rehospitalization for respiratory illness in infants of less than 32 weeks gestation. Pediatrics 1991 ;88: 527-32.
Dagan R.,Tal A. Hospitalization of Jewish and Bedouin infants in Southern Israel for bronchiolitis caused by respiratory syncytial virus. Pediatr Infect Dis J, 1993; 12: 381-6
Elder D. E., Hagan R., French N. P. Hospital admission in the first year of life in very preterm infants. J Paediatr Child Health 1999; 35(2): 145-50.
Furman L., Hack M. Hospitalization as a measure of morbidity among very low birth weight infants with chronic lung disease. J Pediatr 1996; 28: 447-52.
Hewitt M., Brown P., Boston V. Inguinal herniotomy in young infants, with emphasis on premature neonates. J Pediatr Surg 1994;29: 1496-8.
Kitchen M. D., Kelly E. Health and hospital readmissions of very low birth weight and normal birth weight children. AJDC 1990; 144: 213-18.
Mc Cormick M. C., Shapiro S., Starfield B.H. Rehospitalization in the first year of life for highrisk survivors. Pediatrics 1980; 66: 991-99.
2. SECUELAS DE ENTEROCOLITIS NECROTIZANTE
Liliana Bouzas
La Enterocolitis Necrotizante (ECN) es una de las enfermedades más severas del prematuro que ocasiona una mortalidad del 20 al 50% de los niños que la padecen. No sólo es la complicación gastrointestinal más frecuente, sino que muchos de los que sobreviven presentan riesgo de desnutrición, trastornos del crecimiento, complicaciones gastrointestinales, retraso del desarrollo y reinternaciones.
Las secuelas a largo a plazo están relacionadas a la severidad de la enfermedad (I Sospecha, II Enfermedad, III Enfermedad Grave, según los criterios clínicos y radiológicos de Bell).
La mayoría de estos pacientes habrán sido identificados previamente al alta, requerirán ser controlados en forma conjunta con especialistas, pero es necesario que el médico de cabecera conozca las más frecuentes.
Estenosis
El 10% de los niños que padecieron ECN presentarán complicaciones gastrointestinales, dentro de las cuales la estenosis es la más frecuente. Está presente en el 10 al 35% de los sobrevivientes, consecuencia de la reparación y subsecuente retracción cicatrizal de áreas isquémicas que no se han perforado. Aparecen en el intestino distal, aunque la mayoría se manifiesta durante la etapa aguda, otras no se hacen evidentes hasta los 6 meses, después del episodio inicial. Se encuentran estenosis múltiples en 1/3 de los casos.
Hay muchas estenosis que no causan obstrucción y permanecen silenciosas, asintomáticas y sólo se detectan con un examen con enema de bario.
Algunas de las estenosis que se manifiestan en el seguimiento dan síntomas de obstrucción distal lo que incluye dolor, distensión, vómitos y constipación.
La obstrucción puede suceder repentinamente y estar asociada a un cuadro séptico o perforación.
El 5% de los niños que requirieron cirugía tienen un riesgo adicional de desarrollar obstrucción intestinal secundaria a adherencias que aumenta en relación a lo complicado que fue el procedimiento.
|
Al dar de alta a estos niños debe advertirse a los padres acerca de los signos de obstrucción. Ante los signos de alarma (irritabilidad, cólicos, vómitos) deben consultar inmediatamente. Debe obtenerse una Rx de pie o en decúbito supino y lateral para detectar las asas dilatadas y los niveles aire-líquido y consultar al cirujano. El sangrado persistente es frecuente, también requiere ser evaluado mediante enema baritada y derivado a consulta con el cirujano. El sangrado aislado no indica necesidad de reinternación. |
Síndrome de intestino corto
El manejo del síndrome de intestino corto requiere la comprensión de todos los problemas que constituyen el síndrome de diarrea crónica: malabsorción, retraso de crecimiento, deficiencias vitamínicas y minerales.
Se considera la complicación más grave. Se presenta en el 23 % de los niños que requirieron cirugías extensas. La ECN es la causa más frecuente de intestino corto.
El pediatra debe estar al tanto de los cambios anatómicos que se han producido; la extensión del intestino resecado, el lugar de la resección y si la válvula ileocecal ha sido eliminada. Probablemente esto sea, desde el punto de vista funcional, el dato de mayor importancia sea cual fuere la longitud del resecado.
Para que se presenten signos de malabsorción, debe haberse resecado más del 50% del intestino delgado.
El yeyuno es el principal sitio de digestión y absorción de hidratos de carbono. Sin embargo, las resecciones de yeyuno se toleran bien dada la capacidad del íleon de adaptarse y compensar la pérdida de función yeyunal.
Las resecciones ileales se toleran peor dado que allí se encuentran los lugares de mayor absorción de nutrientes particularmente grasas, factor intrínseco ligado a la vitamina B12 y sales biliares conjugadas. Después de la resección ileal se observa aceleración del tránsito intestinal, sobre todo si se eliminó la válvula ileocecal. La válvula ileocecal es una barrera que prolonga el tránsito intestinal.
El colon representa la mayor superficie de absorción de agua y en el neonato funciona como un camino para rescatar hidratos de carbono. La severidad de la diarrea está vinculada a cuanto colon contiguo quedó.
Los cambios fisiológicos intestinales luego de la resección pueden afectar: digestión, motilidad, absorción y secreción, por una disminución del área de absorción, depleción enzimática e hipermotilidad intestinal.
Los niños que sobreviven a ECN son vulnerables a la sobreinfección y a episodios sépticos que amenazan su vida. La sobreinfección puede suceder en un asa aislada, o como consecuencia de la falta de válvula ileocecal que permite a las bacterias colónicas migrar desde el intestino grueso al delgado.
La nutrición parenteral ha permitido grandes avances en el manejo de esta patología. Gracias a ella han podido sobrevivir niños con intestino tan corto como 15 centímetros. Luego del episodio isquémico agudo todos los niños reciben alimentación parenteral. La evidencia ha demostrado que la alimentación parenteral facilita la adaptación intestinal promoviendo la hipertrofia, alargamiento y dilatación del intestino. La transición a la alimentación oral dependerá de esta adaptación y deberá ser muy gradual.
A partir de la segura administración de la alimentación parenteral en forma domiciliaria, algunos niños podrán ser dados de alta antes de haberse completado la transición a la alimentación oral.
Alimentación al alta
Muchos niños con intestino corto pueden tolerar pequeños volúmenes de aportes orales simples. Muchos necesitan fórmulas especiales durante los primeros meses, hasta que la superficie absortiva y las reservas enzimáticas del intestino se hayan restablecido.
Raramente, se necesitan fórmulas especiales sin grasas o carbohidratos. Los substratos se irán agregando a medida que se desarrolle la tolerancia. Lograr nutrición enteral lleva tiempo por lo cual los sobrevivientes de ECN., deben controlarse en cuanto a signos de mala absorción o desnutrición.
Manejo del síndrome de intestino corto
Requiere reconocer los signos específicos de mala absorción: diarrea, distensión, restos alimentarios no digeridos en materia fecal y de desnutrición: aumento escaso de peso, anemia e hipoproteinemia.
La tolerancia a los hidratos de carbono tiene amplias variaciones y está relacionada a la disminución de actividad de las disacaridasas, específicamente lactasa y sacarasa, estando menos afectada la actividad de la maltasa. Esto permite el uso de polímeros de la glucosa-polimerosa como suplementos calóricos. Estos no aumentan la osmolaridad de las fórmulas minimizando el riesgo de diarrea.
Las grasas son las de peor digestión y absorción.
Los triglicéridos de cadena mediana son los mejores tolerados. Una fuente de calorías primarias o suplemento es el TCM (triglicéridos de cadena mediana).
Después de resecciones amplias, las proteínas son el sustrato mejor tolerado. Es mejor administrarlas en forma predigerida. Los oligopéptidos se absorben mejor que los aminoácidos individuales, que requieren un transportador específico.
Todos los niños con "Síndrome de intestino corto" requieren suplementos vitamínicos por largo plazo, especialmente vitaminas liposolubles y duplicar la dosis de vitaminas habituales. El raquitismo es frecuente requiriendo calcio y suplementos adecuados de vitamina D.
Resecciones de intestino proximal requieren hierro y suplementos de folatos.
Aquellos niños que fueron sometidos a resecciones distales ileales necesitaran administración parenteral de vitamina B12. Puede ser necesaria la suplementación de oligoelementos.
La supervisión de un equipo médico clínico y gastroenterológico será necesaria cuando aparezca riesgo de infección o complicaciones metabólicas.
Si bien la hipersecreción gástrica es un fenómeno transitorio, puede requerir tratamiento con cimetidina. En ocasiones, la diarrea acuosa hace necesaria la indicación de colestiramina o resinas intercambiadoras de iones para fijar ácidos biliares no absorbidos. En algunos casos podría estar indicado el uso de agentes antiperistálticos para controlar la frecuencia de deposiciones pero es aconsejable la supervisión de un gastroenterólogo. Cuando se sospecha sobreinfección en un asa, el tratamiento debe ser enérgico pudiendo llegar a ser necesaria la extirpación del segmento.
Cualquier signo de infección sistémica merece tratamiento.
El médico de seguimiento debe aceptar que la recuperación del intestino corto puede ser de meses a años. Dado que el intestino delgado tiene notable capacidad de adaptarse y crecer, muchos pacientes superan el síndrome. La depleción enzimática se va corrigiendo y la superficie de absorción aumenta. Mientras tanto, exacerbaciones y recaídas en el manejo son comunes y esperables. Pocos niños continúan presentando signos después de los dos años. En general, a esta edad logran completar su alimentación vía oral y alcanzan crecimiento compensatorio y la dieta puede acercarse a la del niño sano.
Sin embargo, algunos continúan con intolerancia a ciertos alimentos. Los signos incluyen: diarrea, erupciones, distensión y alimentos no digeridos en materia fecal. Los alimentos más problemáticos son la leche entera, choclo, tomate, jugo de naranja, manzana, durazno, pasas de uva, arvejas y chocolate.
Colestasis
La primera causa de morbilidad tardía es el trastorno hepático colestático causado por la alimentación parenteral prolongada. Se caracteriza por hiperbilirrubinemia a predominio directa mayor a 2 mg/100 ml, hepatomegalia y aumento de transaminasas séricas. Aparece después de la 2ª semana de alimentación parenteral.
Aunque se debe a un conjunto de factores, el más importante es el ayuno prolongado. Es menos claro el papel de las soluciones utilizadas.
El tratamiento más efectivo es iniciar precozmente la alimentación enteral. Esta tiene un efecto trófico sobre la mucosa intestinal lesionada y estimula el flujo biliar.
El volumen no parecería ser de mayor importancia. En el momento del alta deberían haberse descartado todas las otras causas posibles de colestasis y el trastorno estar en vías de resolución. Con el uso de alimentación trófica la mayoría de las colestasis se resuelven entre 1 a 3 meses, escasos pacientes evoluciona hacia la insuficiencia hepática.
Los pacientes que luego del alta continúan con alimentación parenteral o aquellos cuya colestasis no se ha resuelto deben controlarse con evaluaciones periódicas de su función hepática. En ellos el crecimiento se verá afectado a pesar de su adecuado aporte calórico. Si la evolución es desfavorable sus diagnósticos deben ser reevaluados.
Ostomías
Son la exteriorización del intestino a través de la pared abdominal abocándolo a la piel con el objeto de crear una salida artificial al contenido del mismo (yeyunostomía, ileostomía, colostomía) Muchos de los pacientes internados por enterocolitis requieren ileostomías hasta que el episodio isquémico se haya resuelto. La mayoría son luego cerradas antes que el niño se vaya de alta para mejorar la absorción y favorecer la nutrición.
Los niños con intestino corto suelen tener ostomías durante varios meses encarándose su cierre de acuerdo a las razones que las motivaron, al crecimiento que logra el paciente y a las ventajas que le representa la re anastomosis. De ser posible se retrasará el cierre hasta después del año de edad ya que las adherencias pueden aumentar si el cierre se anticipa.
Aquellos que se van de alta colostomizados deben ser especialmente controlados en cuanto a pérdidas que pueden representar un riesgo de vida. No hay que olvidar que el contenido intestinal posee alto porcentaje de proteínas, agua, electrolitos y elementos traza.
La mínima gastroenteritis puede producir rápidamente deshidratación y acidosis. Las pérdidas crónicas de bicarbonato de sodio pueden ser responsables de falta de aumento de peso y deben compensarse a fin de lograrlo.
Cuidado de las ostomías:
Colocación del sistema:
· Realizar la limpieza de la piel que rodea la ostomía con jabón neutro que no deje película aceitosa.
· Secar correctamente.
· Proteger la piel: si ésta se daña no permitirá la colocación de ningún producto ni el cierre de la ostomía.
· Marque en la piel la superficie a cubrir; la abertura que debe adaptarse al sitio de la ostomía debe quedar lo más cercana posible al mismo sin tocar el estoma.
· Cortar la placa de modo que se adapte perfectamente a la superficie marcada.
· Al retirar la antigua bolsa, higienizar con agua caliente. Los baños no están contraindicados.
· Cambiar la bolsa cada tres días o si muestra pérdidas.
Los problemas más comunes:
1) Irritación y sangrado de la piel:
El sangrado leve es frecuente y responde a compresión suave. Deben controlarse pérdidas de sangre oculta.
Si hay irritación el sitio debe permanecer perfectamente limpio. Cubrir el área erosionada con polvo somato adhesivo. Todo el área irritada tiene que estar cubierta por la placa de la ostomía. Es frecuente la complicación por hongos, puede causar una dermatitis que se asemeja a la del pañal. Tratar con polvo con nistatina; ya que las cremas impedirán la adherencia de las bolsas.
Las colostomías ubicadas hacia la derecha se caracterizan porque las deposiciones pueden ser líquidas hacia semi formadas; lo mismo pasa con algunas de las ubicadas en el transverso. Cuando las pérdidas son de alguna consistencia pueden utilizarse para contenerlas pañales en lugar de bolsas.
El ostoma no presenta sensibilidad pero sangra frecuentemente al contacto. En caso de irritación franca quitar la bolsa durante la noche y cubrir, de todas maneras, con la mezcla de polvo adhesivo y pomada protectora permitiendo a la piel airearse.
2) Deshidratación
Puede ocurrir fácilmente en niños ileostomizados aun en ausencia de enfermedad intercurrente. Si ocurren pérdidas mayores a las habituales deben reponerse.
La frecuencia de episodios de deshidratación marca la necesidad del cierre de la ostomía.
3) Obstrucción:
Las ileostomías y ocasionalmente las colostomías pueden obstruirse. Es conveniente instruir a los padres sobre la necesidad de consultar cuando noten ausencia de drenaje durante 12 a 18 horas.
Crecimiento
Al alta, en aquellos niños sobrevivientes a una ECN, la mayoría de los problemas nutricionales y gastrointestinales estarán resueltos, y la mayor parte de los niños podrá lograr buen crecimiento compensatorio.
Muchos de los resultados en seguimientos a largo plazo datan de épocas en que el recurso de la alimentación parenteral no estaba tan difundido.
Si el paciente ha padecido enterocolitis pero no desarrolló un síndrome de intestino corto su crecimiento será comparable al de otros prematuros.
Aquellos pacientes que no hayan superado totalmente su trastorno gastrointestinal pueden necesitar que se mantenga un aporte parenteral o recibir por gavaje durante la noche un apoyo a la alimentación que ya toleran por boca.
La principal meta terapéutica durante el seguimiento será mantener el aporte necesario para conseguir un crecimiento óptimo. El mantenimiento de un crecimiento adecuado mejora el pronóstico de su condición médica y su desarrollo. La experiencia de diversos autores coincide en que cuanto más pequeño y enfermo haya estado el niño mas afectado estará su crecimiento. Es esperable que el 40% de los niños que fueron operados no hayan alcanzado el percentilo 5 para peso a la edad de 4 años. Si bien algunos autores no hallaron diferencias relacionadas con el grado de enfermedad M. Hack en su seguimiento encuentra mayor compromiso entre los niños mas graves, y mayor frecuencia de microcefalia entre los que sobrevivieron a ECN grado III.
Desarrollo
A medida que la sobrevida luego de ECN fue mejorando surgió la preocupación por si la calidad de la misma era adecuada.
Los trastornos del desarrollo observados están relacionados con la prematurez y sus secuelas.
Las complicaciones adicionales que se agregan a la enterocolitis o más aún el intestino corto (desnutrición y malabsorción) repercuten negativamente sobre el desarrollo.
El efecto de la desnutrición sobre el desempeño intelectual está vinculado a la limitación del crecimiento cefálico.
Además, durante la etapa aguda la enterocolitis puede predisponer a meningitis por entero bacterias tras internación prolongada y más aún las reinternaciones por la enfermedad no totalmente resuelta suman su efecto adverso.
En estos niños suele verse una menor velocidad de desarrollo. El retraso motor que muestran a los doce meses generalmente está resuelto a los dos años. Así mismo es más frecuente encontrar comprometida la evolución. El tipo de limitaciones no difiere de las encontradas en otros prematuros.
No es difícil aceptar que estos niños, que atravesaron un fallo multisistémico, tengan retraso más severo. Muchos factores pudieron afectar al sistema nervioso por sí solos o uniendo su efecto: la hipoperfusión que favoreció la ECN ejerció efecto perjudicial sobre la circulación cerebral, el schok, acidosis e hipoxemia generan una encefalopatía hipoxicoisquémica, mediadores citotóxicos liberados durante la inflamación sistémica, sepsis asociada o no a meningitis, desnutrición e hipercatabolismo, limitando el crecimiento y diferenciación neuronal antes del las cuarenta semanas.
Controles
Deben establecerse de acuerdo a las necesidades específicas del niño y su familia. La primera citación debe hacerse entre 1 y 2 semanas para establecer el estado actual de la enfermedad, problemas no resueltos, antropometría, dieta.
La transición del cuidado intensivo al seguimiento se hace especialmente difícil para estas familias acostumbradas a demandar un médico capaz de contenerlos.
Cuando existan problemas de crecimiento los controles deben hacerse cada una a 2 semanas. Las citaciones frecuentes permiten continuar la educación de estos padres, darles signos de alarma que sirvan como prevención de reinternaciones.
BIBLIOGRAFIA
Bernbaum-Williamson J. Gastrointestinal issues. Primary care of the preterm infant. St. Louis, Mosby Year Book, 1991.
Howell L. Home Ostomy care. in Ballard: Pediatric of the ICN Graduate. Philadelphia, Saunders, 1988.
Ladd A. Rescoria F. Grosfeld J. Long term follow-up after Bowel resection for necrotizing enterocolitis: Factors affecting outcome J Pediatric Surgery,1998; 33: 967-972.
Lorimer A. Care of ICN Graduates after neonatal surgery. In Ballard: Pediatric of the ICN Graduate. Philadelphia, Saunders, 1988.
Simon N. Follow-up for infants with necrotizing enterocolitis. Clinics Perinatology, 1994;21(2): 411- 424.
Sontag J., Grimer I. Growth and neurodevelopmental outcome of very low birthweight infants with necrotizing enterocolitis. Acta pediátrica, 2000; 89: 528-32.
Walsh M., Kliegman, Hack M. Severity of necrotizing enterocolitis influence on outcome at 2 years of age. Pediatrics, 1989; 84 (5).
3. ENFERMEDAD OSEA DEL PREMATURO
Patricia Climent
Definición:
La enfermedad metabólica del hueso es un trastorno frecuente de los niños pretérminos menores de 32 s. y 1500 g al nacer. Se caracteriza por un déficit de calcio y fósforo que produce inadecuada mineralización del tejido osteoide e incluye perturbaciones que van desde la hipomineralización leve (osteopenia) hasta la enfermedad ósea grave con fracturas (raquitismo). La osteopenia se manifiesta en la etapa de crecimiento rápido, alrededor del 2º o 3º mes de vida postnatal. El raquitismo (displasia epifisaria y deformidades esqueléticas) se observa después de los 2 a 4 meses de edad corregida.
Incidencia
La incidencia de la enfermedad ósea metabólica depende de la edad gestacional, el peso, el grado de enfermedad neonatal, el uso de alimentación intravenosa u oral, la ingestión o el aporte intravenoso de minerales y el uso de diuréticos o corticoides. La enfermedad ósea del prematuro se observa en más del 30% de los pretérminos menores de 1500 g y en el 50% de los menores de 1000 g.
Etiología
Durante el último trimestre de la vida intrauterina se produce una rápida mineralización ósea, con el depósito del 80% del contenido mineral que el niño tendrá al término. En el útero los valores de acreción mineral aumentan en forma exponencial desde la semana 24 a la 36 inclusive, con un ritmo de mínimo de 120 –150 mg de Ca/Kg/día y máximo de 150-210 mg de Ca/Kg/día y de 75 a 140 mg de P/Kg/día. Estas necesidades son cubiertas por un transporte activo de Ca y P a través de la placenta. La brusca separación de su fuente de minerales a las 24 a 32 semanas de gestación tiene un gran impacto sobre la mineralización ósea.
La mineralización ósea requiere valores adecuados de Ca y P en el compartimento extracelular y actividad osteoblástica normal. El lactante de muy bajo peso al nacer duplica el peso en 2,5 meses, por lo que cualquier anomalía en la ingestión, disminución de la absorción gastroenteral o eliminación anormal de Ca o de P tendrá por consecuencia una mineralización ósea insuficiente.
A) DEFICIT DE CALCIO Y DE FOSFORO
a.1) Reservas minerales disminuidas
• Prematurez (en el último trimestre se deposita el 80% del contenido mineral óseo).
• RCIU (la insuficiencia placentaria implica menor aporte de Ca y P).
a.2) Aportes insuficientes
La nutrición parenteral total prolongada contribuye a la mineralización ósea deficiente debido a que el aporte de Ca y P en las soluciones parenterales, la cantidad y calidad de los aminoácidos, el pH de la solución, la Tº de almacenamiento y el orden de mezcla de los minerales son factores que afectan la solubilidad del Ca y el P parenteral. El uso de soluciones con aportes de Ca de 20 a 32 mg/100ml, y de P de 15 a 25 mg/100ml ha causado importante enfermedad ósea y alteraciones bioquímicas. El uso de soluciones con 60 a 80 mg/100ml de Ca y 47 a 62 mg/100ml de P no ha condicionado el desarrollo de osteopenia. La relación Ca / P recomendada por la mayoría de los investigadores varía entre 1,3:1 y 2:1, en un intento de conservar la relación fisiológica.
La leche humana, según la mayoría de los autores, tiene un aporte insuficiente de Ca, P, y vitamina D. En un lactante que recibe 200 ml/Kg/d proporciona 51 a 68 mg de Ca /Kg/d y 21 a 28 mg de P/Kg/ d, por lo que no podría cubrir las necesidades del niño pretérmino. Por lo tanto requeriría de polvo enriquecedor hasta que el pretérmino logre un peso de 1800 a 2000g. Algunas comunicaciones refieren que la leche materna, a pesar de su bajo aporte, tiene factores adicionales que permiten mejor absorción y metabolización de estos minerales y a largo plazo los niños lograrían una mineralización ósea adecuada y similar a niños con leches suplementadas.
a.3) La excreción exagerada de Calcio
La enfermedad renal crónica o el aporte de Furosemida producen pérdidas urinarias prolongadas.
a.4) La absorción deficiente de Calcio y malabsorción de grasas.
La enfermedad hepática (colestasis), gastroenteral o renal prolongadas pueden afectar la absorción y/o la metabolización y activación de la vitamina D (liposoluble).
B) DEFICIT DE VITAMINA D
No es la causa principal. Se puede deber a:
b.1) Déficit de aporte
b.2) Insuficiente hidroxilación hepática de la vitamina D
En prematuros inmaduros parece ser insuficiente la hidroxilación hepática de la Vitamina D, por lo que la administración de 400 UI de vitamina D podrían ser insuficientes, sugiriendo algunos autores la necesidad de 1000 UI de vitamina D en niños menores de 1000g por falta de respuesta del tejido óseo a la vitamina D activada. Sin embargo esto está discutido y Tsang ha demostrado que el contenido de las fórmulas de prematuros, con aportes tan pequeños como 160 UI/día sería suficiente y mantendría estable las concentraciones séricas de 25-OHD y 1,25(OH)2D por lo que aconseja una dosis de 400 UI/día.
b.3) Déficit de absorción de grasas y vitaminas liposolubles (colestasis)
b.4) Medicamentos como el Fenobarbital interfieren la hidroxilación hepática.
DIAGNOSTICO
a. Signos clínicos
Las manifestaciones clínicas incluyen craneotabes, que puede llegar a palparse en todo el cráneo, rosario costal, fracturas patológicas, que por lo general se presentan después de los 2 ó 3 meses de vida postnatal y corresponden a una edad postconcepcional de 38 a 42 semanas.
La reducción o detención en el crecimiento de la longitud corporal es un dato clínico temprano y sumamente valioso de enfermedad ósea.
b. Laboratorio
El control de laboratorio puede alertar cuando la osteopenia es subclínica. Los análisis de rutina en seguimiento muestran:
• Calcemia (Ca) y magnesemia (Mg) normales,
• Hipofosfatemia (P) bajos (<4.5 mg %) o normales
• Aumento de actividad de fosfatasa alcalina en suero (> 450 UI/dl), en el prematuro en crecimiento se aceptan como normales valores de hasta 5 veces los del adulto (90-260 U/l): hasta 1076U/litro.
Tabla Nº 7:
|
Etiología de la enfermedad ósea del Prematuro CAUSA PRINCIPAL: DEFICIT DE CA Y P: a) Reserva mineral inadecuada: Prematurez RCIU b) Aportes insuficientes en la etapa neonatal Nutrición parenteral Leche materna no suplementada Fórmulas no destinadas para prematuros c) Absorción insuficiente de Ca y P (mala absorción, jabones cálcicos) d) Pérdida renal de Calcio y fósforo e) Medicamentos: Furosemida o corticoides (Broncodisplasia) Otra causa: Déficit de Vitamina D: a) Déficit materno b) Ingesta o absorción inadecuada (ECN, resección intestinal) c) Enfermedad hepatobiliar (colestasis neonatal) d) Insuficiencia renal crónica e) Administración de fenobarbital (aumenta el metabolismo de 25(OH)D |
• En casos especiales podrían solicitarse:
• Hormona paratiroidea (PTH ):
Valores normales o aumentados • 25 OH vitamina D (25-OHD): concentración sérica baja o normal.
• 1,25 dihidroxivitamina D (calcitriol), aumentado.
La elevación de la actividad de la fosfatasa alcalina (FA) puede detectarse entre las 3 a 4 semanas postnatales
• 1) Control de actividad de fosfatasa alcalina es un indicador valioso de enfermedad ósea.
• 2) El nivel de fósforo sérico bajo alerta sobre baja mineralización ósea.
• 3) El nivel de calcio sérico (generalmente normal) no es orientador.
c. Radiología
Los cambios radiográficos se manifiestan después de las 4 a 6 semanas de edad postnatal e incluyen:
• desmineralización ósea difusa
• metáfisis irregulares
• formación subperióstica de hueso nuevo
• epífisis ensanchadas y desflecadas
• fracturas patológicas
PREVENCION Y TRATAMIENTO
Debemos tener en cuenta que el prematuro tendrá:
• Pequeñas reservas de Ca y P, las que serán más severas a menor edad gestacional por falta de aporte placentario en el 3º trimestre.
• Absorción deficiente de minerales y vitaminas liposolubles comparadas a la transferencia placentaria.
• Inmadurez hepática en los muy inmaduros para almacenamiento de vitamina D como 25OHD.
• Inmadurez renal para la reabsorción de Ca y P y la activación de la vitamina D a calcitriol.
Controles de laboratorio
Es necesario monitorizar periódicamente calcemia, fosfatemia y fosfatasas alcalinas, para detectar la elevación o el descenso de las FA así como prevenir la hipercalcemia.
Aportes de calcio y fósforo
La prevención es más fácil que el tratamiento. El aporte de Ca y P debe iniciarse precozmente aportando parenteralmente el máximo que permita la solución sin precipitación, luego continuar con leche de madre fortificada o leche de fórmula para prematuros.
El aporte de minerales debe continuarse por 8 a 10 semanas hasta lograr un peso de 2000 a 2.500g (aproximadamente hasta las 40 s), podría prolongarse en los pretérminos que requirieron alimentación parenteral prolongada y en los muy inmaduros. La absorción de calcio es del 60 +/ - 15 % de lo administrado y la de fósforo es del 70%.
La Academia Americana de Pediatría, Comité de Nutricion (AAP-CON) aconseja un aporte de 185 mg/Kg/d de Ca, 123 mg/Kg/d de P y 8,5 mg/Kg/día de Mg y 500 UI/día de Vitamina D.
La Sociedad Europea de Pediatría, Gastroenterología y Nutrición (ESPGAN) aconseja 128 mg/ Kg/d de Ca, 70 mg/Kg/d de P y 7 mg/Kg/d de Mg y 800 a 1600 UI/día de Vitamina D.
Las leches de prematuros oscilan entre 80 y 146 mg de Calcio y 60 a 90 mg de Fósforo cada 100 ml de fórmula reconstituida al 15% (Ver Anexo Nº 8) Los sucedáneos de la leche materna aportan entre 40 a 60 mg de Ca y 25 a 60 mg de Fósforo.
La leche humana puede ser enriquecida con fortificadores que aportan: en 2 g 45 mg de calcio y 22.5 mg de Fósforo, se diluye en 50 o 100 ml según el cálculo de aportes.
Los suplementos en forma de comprimidos de lactato de Ca de 500 mg (65 mg de Ca elemental), pueden fraccionarse en sellos de 250 mg y administrarse repartidos en 2, 3 o 4 tomas. El fosfato monopotásico o dipotásico puede utilizarse.
Deben controlarse el aporte cuidadosamente para evitar errores de administración. No se aconseja su administración el biberón por el riesgo de precipitación.
En los niños que reciben diuréticos o corticoides el aporte elevado de calcio puede favorecer el desarrollo de nefrocalcinosis.
Debería reemplazarse el uso de furosemida por diuréticos tiazídicos.
Vitamina D
En pretérminos se aconsejan entre 400 y 600 UI diarias. En los niños menores de 1000g al nacer o con alimentación parenteral prolongada, entre 400 y 1000 UI diarias.
Los sucedáneos de la leche materna aportan alrededor de 40 UI cada 100 ml al 12.6%.
Las leches de prematuros aportan aproximadamente 58 UI cada 100 ml al 15%.
Los fortificadores de leche materna aportan 150 UI cada 2g. (Ver Anexo Nº 8).
Tabla Nº 8: Distintas Recomendaciones de Ca, P y Vitamina D
|
. |
TSANG Y COL. |
AAP-CON (1) |
ESPGAN-CON(2) |
NC-CPS(3) |
|
Calcio (mg) |
120-230/Kg/día |
185/Kg/día |
70-140/Kg/día |
160-240/Kg/día |
|
Fósforo (mg) |
60-140/Kg/día |
123/Kg/día |
50-90/Kg/día |
77,5-118/Kg/día |
|
Relación Ca/P |
2:1 |
. |
. |
1.6-2:1 |
|
Vitamina D |
150-400 UI/día |
400 UI/día |
800-1600 UI/día |
400 (800) UI/día |
1) Academia Americana de Pediatría, Comité de Nutrición 2) Sociedad Europea de Pediatría, Gastroenterología y Nutrición 3) Comité de Nutrición, Sociedad Canadiense de Pediatría
MANEJO PRACTICO
a. Suplemento preventivo
• Pretérminos sanos
Pretérmino mayor de 1500g: no requiere suplementar Ca y P de rutina. Controlar laboratorio a las 40 s y a los 2 meses corregidos. Aportar vitamina D a 400 UI/día.
Pretérmino menor de 1500g: suplementar Ca y P de rutina hasta las 40 semanas. Control de laboratorio a las 40 semanas, a los 2 y 5 meses. Administrar vitamina D 400 UI/día. En el menor de 1000g evaluar la necesidad de dosis mayores de Vitamina D.
• Pretérminos de cualquier peso, enfermos o con riesgo elevado de osteopenia-raquitismo: suplementar hasta finalizar el crecimiento rápido Dosis de suplemento preventivo:
Calcio 100-130 mg/Kg/día
Fósforo 50-65 mg/Kg/día
Vitamina D 400 UI/día
Controlar calcemia, fosfatemia y fosfatasa alcalina mensualmente mientras reciban suplemento. Si la calcemia aumenta de 10 mg% o la fosfatemia de 6 mg% suspender el suplemento.
b. Tratamiento:
Si existen signos clínicos o radiológicos de osteopenia o raquitismo y si la Fosfatasa alcalina fuera > de 1076/l y la fostatemia < de 4,5 mg%.
Aumentar aporte de Ca a 200 mg/Kg/día.
Aumentar aporte de P a 100 mg/Kg/día.
Aumentar aporte de vitamina D a 1000 UI/día.
Controlar cada 2 semanas calcemia, fosfatemia y fosfatasas alcalinas para controlar la evolución. Mantener el tratamiento hasta lograr crecimiento adecuado y normalización del laboratorio.
BIBLIOGRAFIA
Azrilevich E., Perasso V. Normas de seguimiento ambulatorio del niño con bajo peso al nacer, menor de 2500 g. Neuquén, Subsecretaría de Salud, Provincia de Neuquén, 1995.
Bishop N. J., Dhalenburg S.L., Lucas A. Dieta temprana en el pretérmino y mineralización ósea a la edad de cinco años. Acta Pediátrica Escandinava, 1996; 85: 230-6.
Bishop K. L.. Enfermedad ósea en prematuros. Arch. Dis. Child 1989 Bishop, K. L. Increased bone mineral content of preterm infants fed with a nutrient enriched formula after discharge from hospital. Arch of Dis in Child 1993;68:573-578
Cloherty J. Stark A. Manual de cuidados intensivos neonatales 3ª Ed Boston, Masson., 1999: 635- 7
Koo W. W. , Tsang R. C., Krug-Wispe S. Effect of three levels of Vitamin D Intake in preterm infants receiving high mineral–containing milk J. Pediatric Gast Nutr, 1995; 21:182-189
Nutrition Comittee Canadian Paediatric Society. Nutrient needs and feeding of premature infants. Can Med Assoc,1995;152 (11) 1765-1785
Schanler R. J. and Rifkca R. Calcium, Phosporus and Magnesium needs for the low birth-weight infant. Acta Paediatr Scan Suppl 405:111- 16.1994
Toeusch W., Yogman M. W. Follow-up management of the high-risk infant. Boston/Toronto, Little, Brown and Company, 1987:196-199
Tsang, N. Nutrition during infancy. Philadelphia, Hanley-Belfus,1988
4. EVENTOS DE AMENAZA APARENTE A LA VIDA
(Apparent Life Threatening Event: ALTE)
Alejandro Jenik
DEFINICION
Se define ALTE como un episodio que alarma al observador y que está caracterizado por alguna combinación de pausa respiratoria, cambio de color (cianosis, palidez o rubicundez), cambio en el tono muscular (usualmente hipotonía), ahogos o arcadas. En algunas ocasiones, el observador experimenta la sensación de estar ante una muerte inminente. El evento puede revertir espontáneamente, requerir estimulación vigorosa o resucitación cardiopulmonar.
En el pasado se utilizó el término "Muerte Súbita Frustrada" para definir estos episodios; pero el Consenso de Apnea Infantil y Monitoreo Domiciliario del año 1986 en los Estados Unidos, sugirió abandonar este último término debido a que una pequeña proporción de lactantes fallecidos con el diagnóstico de Síndrome de Muerte Súbita del Lactante (SMSL) tiene historia previa de apnea, cianosis o ALTE (7% en los datos presentados por el Instituto Nacional de Salud de los Estados Unidos y 8,8% en la serie Australiana) y para la gran mayoría la muerte es la primera y única señal.
CARACTERISTICAS DEL ALTE:
Subjetividad del observador:
El diagnóstico de ALTE se basa en la observación realizada por una persona que no tiene entrenamiento médico (padres, abuelos, cuidadores). La presencia o ausencia de movimientos respiratorios, el cambio de color y de tono, como así también la magnitud de la intervención realizada pueden estar muy distorsionados por el susto de la persona que observó el episodio. A veces es difícil diferenciar eventos de ALTE de episodios de ahogo y regurgitación, en los cuales la intervención se realiza muy rápida, quedándonos la duda de si realmente se requería la misma. Muchas veces, "eventos fisiológicos normales", pueden producir una sobre reacción en los padres, particularmente aquellos muy ansiosos o con disturbios psicológicos.
MORTALIDAD DE NIÑOS CON ALTE:
La mayoría de los niños presentan un solo episodio del cual sobreviven con un desarrollo neurológico normal. Se debe informar a las familias de niños con episodios leves y únicos que la incidencia de nuevos episodios es extremadamente baja. Sin embargo, existe un pequeño e infrecuente subgrupo de niños con ALTE, en el cual la mortalidad es elevada:
• Niños con episodios severos y recurrentes que requirieron resucitación cardiopulmonar (RCP), particularmente asociados con epilepsia o historia de SMSL en hermanos anteriores.
• Niños con episodios durante el sueño que requirieron alguna forma de RCP.
• Grupo de pacientes con infección respiratoria y apneas.
• Prematuros con episodios de ALTE.
|
Tabla Nº 9: Distintas situaciones fisiopatológicas que pueden expresarse con ALTE. • Respiratoria Infección (VSR, Pertussis, Clamydia, neumonía). Obstrucción de la vía aérea superior (retrognatia). Obstrucción de la vía aérea inferior (traqueo-broncomalacia). Cortocircuito intrapulmonar (episodios de crisis del sollozo). • Neurológica Convulsiones. Hemorragia endocraneana (déficit de vit K, maltrato). Hipoventilación central (congénita, drogas, enfermedad neurológica). Enfermedad neuromuscular. • Anemia • Disfunción autonómica. • Sobrecalentamiento. • Cardíaca: alteraciones del ritmo cardíaco-cardiopatías congénitas. • Infección: meningitis. sepsis, gastroenteritis, infección urinaria. • Reflujo gastroesofágico (RGE). • Abuso: envenenamiento, intento de sofocación, simulación. • Iatrogénico: drogas, postanestésico. • Errores congénitos del metabolismo • Hijo de madre drogadicta. • Idiopático o Apnea de la infancia: no puede demostrarse patología asociada |
ETIOPATOGENIA
El ALTE no es un diagnóstico en sí mismo sino una forma de presentación clínica (ver Tabla 9).
¿Cómo se estudia un niño con ALTE?
La variedad y complejidad de las posibles causas de ALTE, junto a las dificultades en su manejo hacen necesario un trabajo médico interdisciplinario que permita un enfoque más amplio y objetivo para evitar el riesgo de sobrestimar o subestimar algún diagnóstico.
Todo paciente que presente un episodio de ALTE debe internarse.
La observación de nuevos episodios y un examen físico exhaustivo por el equipo médico pueden ayudar a clarificar la severidad y causa del episodio. Además, la familia luego del episodio puede estar extremadamente ansiosa y la internación ofrece un espacio de contención psicológica y eventuales consejos futuros.
Sin embargo, para los niños que requerieron una mínima intervención o estimulación, presentan un examen físico normal y no tienen evidencias de estar cursando una infección, cabe la posibilidad de no internación.
Monitoreo de las funciones vitales del paciente:
Con la finalidad de prever la recurrencia de nuevos episodios que se producen mayoritariamente durante los primeros días luego de producido el evento. Lo más efectivo es el monitoreo simultáneo de la frecuencia cardíaca y la oxigenación a través de la medición no invasiva de la saturación de oxígeno, utilizando un sensor que se ubica en la mano o en el tobillo del niño. De no contar con un saturómetro para el monitoreo del paciente, se puede optar por un monitor de 2 canales (frecuencia respiratoria por impedanciometría y frecuencia cardíaca), o simplemente por un monitor de frecuencia cardíaca, o eventualmente por la observación de enfermera, madre.
Historia Clínica (es el procedimiento de diagnóstico más importante).
Detallada historia del evento:
¿Qué color tenía la cara y el cuerpo del niño? ¿Estaba despierto o dormido? ¿Cuánto tiempo duró el episodio? ¿Qué intervención se realizó para abortarlo? ¿Tuvo relación con la comida, posición, llanto? ¿Tenía movimientos anormales? ¿Estaba febril? ¿Quiénes estaban presentes durante el mismo? ¿Dónde ocurrió el mismo? ¿Cómo era el estado de conciencia luego del episodio?
|
La presencia de sangre en la boca o nariz debe hacer sospechar la posibilidad de sofocación en el niño que presenta un episodio de ALTE. |
Antecedentes maternos: cigarrillo, alcohol, drogadicción.
Antecedentes del niño: ¿Niño vomitador o regurgitador?, Semiología durante el sueño: ronquidos, transpiración, pausas respiratorias. Hábitos durante el sueño: colecho, cohabitación, posición para dormir en el momento del hecho.
Técnica alimentaria.
Examen físico completo con evaluación neurológica
EXAMENES COMPLEMENTARIOS AL INGRESO:
• Bicarbonato sérico: la acidosis metabólica puede indicar una historia de ALTE severo y/o alteración metabólica.
• Hemoglobina / Recuento de glóbulos blancos y fórmula
• Glucemia.
• Orina completa
• Radiografía de Tórax.
• Electrocardiograma.
• Considerar: estudios virológicos, bacteriológicos, electrolitos en sangre y electroencefalograma.
¿Cuándo sospechar una enfermedad metabólica?
Cuando el paciente con ALTE presenta acidosis metabólica se debe solicitar un dosaje de amonio. Se debe sospechar alteración metabólica en un paciente con ALTE cuando el mismo presenta alguno de los siguientes criterios: ALTE recurrente, historia familiar de SMSL/ALTE, hepatomegalia, hiperamoniemia, hipoglucemia o convulsiones.
|
No hay ninguna prueba de laboratorio que nos confirme inequívocamente que el niño presentó un episodio de ALTE. |
¿Cuándo se deben realizar exámenes complementarios de mayor complejidad?
Estudios adicionales (previa discusión interdisciplinaria) se deberían llevar a cabo exclusivamente si de la historia clínica, el examen físico, los resultados de los análisis iniciales, la observación y/o el monitoreo de los signos vitales durante las primeras 24-48 horas en el hospital surge la sospecha de un diagnóstico específico, o si la historia es inusual en términos de la severidad del episodio, recurrencia del mismo o historia familiar.
|
Si la evaluación inicial es negativa y el paciente está estable desde el punto de vista cardíaco y respiratorio, es apropiado diferir adicionales pruebas diagnósticas y supeditar las mismas al curso clínico subsecuente del paciente. |
Los estudios publicados y la experiencia clínica nos indican que si el paciente no muestra eventos durante la internación, las posibilidades que repita apneas y/o bradicardia en la casa son escasas. En contraste, cuando se identifican eventos significativos en el hospital (cianosis, bradicardia, desaturaciones) las posibilidades que repita los mismos en el hogar son muy elevadas.
¿Cuándo se debe solicitar una polisomnografía o neumografía? Si los antecedentes (historia familiar previa de SMSL y/o ALTE) o la historia clínica del paciente y/o su evolución durante la internación sugieren la posibilidad de un episodio convulsivo, apneas obstructivas (sueño con ronquido, profusa sudoración e inquietud), hipopnea con hipoxia durante el sueño, o sospecha de cortocircuito intrapulmonar debemos solicitar una polisomnografía o una evaluación respiratoria con una neumografía según el criterio clínico y la gravedad del episodio.
La neumografía es un estudio que, a través de 4 canales de monitoreo (frecuencia cardíaca, frecuencia respiratoria por impedanciometría, flujo aéreo nasal y saturometría), brinda información necesaria durante un período prolongado (2 a 24 horas) sobre el estado cardiorespiratorio: aparición de apneas, categorización (centrales, obstructivas y mixtas), duración y repercusión cardíaca de las mismas, respiración periódica, y desaturaciones (asociadas a pausas).
El estudio polisomnográfico con oximetría digital, si bien su duración es inferior a la de la neumografía, brinda datos similares más el registro electroencefalográfico. Se debe realizar en lo posible durante la noche, con una duración de sueño espontáneo de por lo menor 6 horas y en presencia de un observador médico o técnico entrenado.
¿Qué evalúa la polisomnografía?:
EEG (sólo en los casos en que cuente con por lo menos 6 canales de EEG izquierdos y 6 derechos se puede valorar la presencia de grafoelementos anormales), alteraciones de la estructura interna del sueño, maduración bioeléctrica (ej. persistencia de "trace alternat") y de las reacciones del despertar.
Además de valorar las alteraciones de la frecuencia cardíaca y de la respiración como en la neumografía, puede valorar la CO2 por capnografía para el diagnóstico de hipoventilación central.
Eventos significativos durante los estudios polisomnográficos o neumográficos
• Apneas obstructivas (ausencia de flujo aéreo con presencia de esfuerzo respiratorio): mayor a 3 segundos de duración.
• Apneas centrales prolongadas (ausencia de flujo aéreo y esfuerzo respiratorio): > de 20 segundos de duración.
Apneas centrales patológicas menores de 20 segundos asociada a bradicardia y/o desaturaciones Apneas centrales cortas (menores de 15 segundos) pueden considerarse normales a cualquier edad.
Bradicardia: menor de 80 latidos por minuto durante el primer mes, menor de 70 latidos por minuto durante el 2º mes, menos de 60 latidos por minuto durante el 3º y 4º mes y menos de 50 latidos por minuto en los lactantes mayores de 5 meses.
Desaturaciones: Línea de base de SpO2 menor del 95% o episodios de SpO2 menor de 80% por más de 4 segundos deben ser considerados anormales, siempre en el contexto del niño.
Rango normal de la saturación de oxígeno en los lactantes:
|
Saturación de oxígeno % |
|
|
Mediana |
97.9 |
|
Percentilo 10 |
95.2 |
Una medición continua de la saturación de oxígeno menor de 95% mientras el niño respira con ritmo y amplitud regular debe ser considerada anormal.
Caídas ocasionales de la SpO2 durante menos de 4 segundos de duración deben ser consideradas normales durante los primeros 6 meses de la vida y coinciden mayoritariamente con episodios de respiraciones periódicas.
El 95 percentilo para la frecuencia de desaturaciones prolongadas (SpO2 menor de 80% por más de 4 segundos) es 3 por 12 horas de registro.
Los prematuros con enfermedad pulmonar crónica pueden presentar hipoxemia no detectada, la cual expone a estos niños a mayor riesgo de episodios hipoxémicos, ALTE y SMSL.
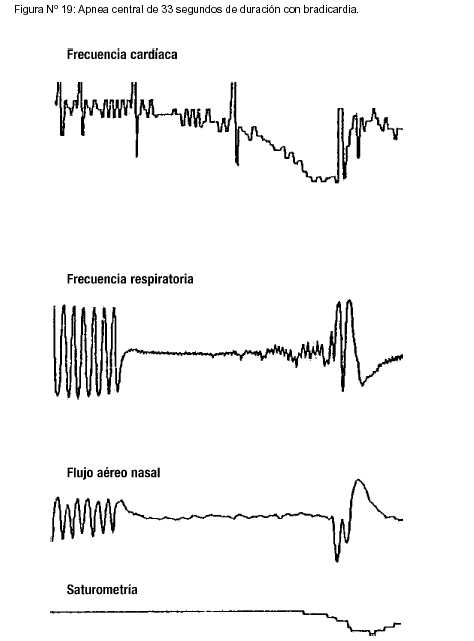
|
Ni la neumografía ni la polisomnografía tienen la suficiente sensibilidad ni especificidad para ser usados prospectivamente para la identificación de pacientes con riesgo futuro de ALTE o SMSL. La imposibilidad de realizar estos estudios durante la internación no debe ser motivo para postergar la externación de estos pacientes, excepto en muy situaciones muy puntuales. |
TRATAMIENTOS ESPECIFICOS:
Reflujo gastroesofágico (RGE):
Continua siendo controvertido si el RGE es la causa de episodios hipoxémicos, un gatillo en niños susceptibles, o de hecho una manifestación asociada. La mayoría de los niños con RGE significativo no experimentan ALTE, por lo que el establecer el diagnóstico de RGE en un paciente con ALTE no demuestra que ésta sea la causa del ALTE, excepto si se logra demostrar temporalmente la asociación entre el evento y el pico de acidez con la pHmetría. Hay diferentes exámenes para diagnosticar el RGE y/o sus efectos, de los cuales la pHmetría esofágica de 24 horas es el de mayor utilidad, aunque en la actualidad se ha descartado este estudio como "patrón oro".
La hipoxemia y la apnea pueden ocurrir en asociación con la alimentación pero sin evidencias de RGE.
Enfoque global del paciente con RGE:
1. Estricta prohibición de exposición al tabaco.
2. Farmacoterapia:
• Agentes prokinéticos:
Publicaciones recientes, año 2000, de los máximos expertos en el tema del RGE, sostienen que debido a la favorable ecuación entre el riesgo y el beneficio del cisapride, el mismo es la medicación de elección para el tratamiento del RGE.
Es ético tratar a niños con evidencia clínica clara de RGE patológico con drogas proquinéticas (ej. cisapride) cuando por algún motivo no se puede realizar la pHmetría.
El cisapride es una droga segura, pero puede estar asociada con serios efectos colaterales si se usa en forma inapropiada. Se conoce el hecho de que el cisapride prolonga el intervalo QTc. Sin embargo, a dosis terapéuticas en niños no hay una asociación directa entre la concentración sérica de cisapride y la prolongación el QTc. De todas maneras se aconseja realizar un ECG previo y luego de 2 ó 3 días de comenzada la administración de la droga.
Situaciones que pueden predisponer a la prolongación del QTc deben ser evitadas: hipokalemia, hipomagnesemia y los siguientes medicamentos: ketoconazol, itraconazole, fluconazol, micomazol, eritromicina, claritromicina, astemisol, tioridazina, y halopurinol (Es importante alertar a los padres y también a los médicos la prohibición de usar concurrentemente estos medicamentos).
La dosis standard es de 0.2 mg/kilo/dosis: 4 veces por día. No debería usarse a una dosis mayor a 0.8mg/kilo/día.
Para los pacientes que no pueden ser tratados con cisapride o no se benefician con la droga, la metroclopramida y el betanecol pueden ser alternativas válidas.
Seguramente en un futuro cercano, podremos contar con drogas proquinéticas con menos efectos colaterales potenciales que el cisapride.
• Agentes inhibidores de la secreción ácida:
Los pacientes pediátricos con sospecha o diagnóstico de esofagitis deben ser tratados con agentes proquinéticos e inhibidores de la secreción ácida (ranitidina, cimetidina).
3. Posición para dormir:
Los niños con diagnóstico de RGE deben dormir en posición plana y en decúbito lateral izquierdo. Se desaconseja la posición "semisentado" debido a que en esta posición el aumento de la presión abdominal contribuye a incrementar el RGE y por otro lado, cuando un lactante pequeño está en esta posición, su cabeza tiende a caer hacia delante con el mentón tocando el tórax lo cual puede restringir la entrada de aire a la vía aérea. En algunos niños, en esta posición, el peso de la cabeza es suficientemente importante como para que la mandíbula se desplace hacia atrás pudiendo producir obstrucción a la entrada de aire.
ALTE Idiopático o Apnea de la infancia:
Existen 3 alternativas terapéuticas: la no intervención, el uso de estimulantes respiratorios (xantinas) o el monitoreo domiciliario. Las xantinas disminuyen el tono del esfínter esofágico inferior, pudiendo empeorar un posible RGE. A nivel del SNC disminuyen el umbral para las convulsiones. Debido que tanto el RGE como los episodios convulsivos se describen en pacientes con ALTE, el médico debería estar razonablemente confiado que estas condiciones no están presentes antes de indicar xantinas. Si los episodios de ALTE se incrementan luego de la introducción de este medicamento, el RGE y/o episodios ictales deberían ser reconsiderados.
PROGRAMA DE EGRESO DOMICILIARIO:
• Control de los factores de riesgo para el Síndrome de Muerte Súbita del Lactante.
• Tratamiento específico si lo hubiere y según el caso
• Curso de Resucitación Cardiopulmonar para padres/cuidadores
• Enlace a sistema de emergencia/centro de derivación
• Monitoreo domiciliario.
Monitoreo domiciliario:
La familia con un niño que requiere monitoreo domiciliario debe participar en un programa que incluya atención médica, técnica y soporte psicológico.
¿Qué pacientes requieren monitoreo domiciliario?
a) Pacientes con ALTE durante el sueño que requirieron reanimación boca a boca.
b) Episodio severo de ALTE sin diagnóstico o recurrente.
c) Episodio de ALTE en hermano de lactante fallecido por el SMSL.
d) Niño con apneas del prematuro no resueltas en el momento del alta,
e) Hermano de dos o más niños fallecidos de SMSL.
f) Niño con displasia broncopulmonar y oxígeno suplementario en el hogar.
g) Niño menor de 1 año traqueostomizado.
¿Qué pacientes no requieren monitoreo domiciliario?
Los recién nacidos normales.
Los prematuros asintomáticos dados de alta, independientemente de la edad gestacional y/o del peso de nacimiento.
Recién nacido con un solo hermano fallecido de SMSL.
¿Cuáles deben ser los límites de las alarmas?
La alarma de pausa respiratoria debe ser 20 segundos y la de bradicardia 70 latidos por minuto. La alarma de taquicardia no es importante.
¿Qué deben conocer los médicos acerca del monitoreo domiciliario?
Sólo hay anecdóticas evidencias de que el monitoreo domiciliario puede disminuir la mortalidad debido a que no se han efectuado estudios controlados y aleatorizados al respecto.
Por el contrario sí esta demostrado que el monitoreo produce grandes inconvenientes:
Limitación de la persona que cuida al bebe de movilizarse con el mismo.
Cambio de estilo de vida en la familia con potencial fraccionamiento del sueño de los padres como consecuencia de la elevada incidencia de falsas alarmas que producen los aparatos de monitoreo domiciliario.
Aproximadamente 2/3 de las alarmas se deben a pérdida de señal del monitor.
¿Cuál monitor es el más apropiado?
La observación de los padres es, en última instancia, la única forma de asegurarse que el niño está "bien". Por lo que en algunas circunstancias se sugiere la ayuda de una enfermera y/o cuidadora nocturna, para que los padres puedan descansar.
No existe el monitor domiciliario ideal. Se prefieren los monitoreos con memoria, para poder interpretar los eventos en los pacientes. El saturómetro podría tener alguna ventaja sobre el monitor de frecuencia cardíaca/respiratoria debido a que los estudios en niños fallecidos por SMSL muestran hipoxemia como el evento inicial y luego una marcada bradicardia muy difícil de revertir a pesar de vigorosas maniobras de resucitación cardiopulmonar. Sin embargo, la elevada cantidad de falsas alarmas que produce el saturómetro en el hogar, lleva a que los padres en alguna oportunidad, discontinúen el monitoreo en sus niños.
¿Cuándo debe suspenderse el monitoreo?
La decisión de la suspensión del monitoreo domiciliario debe basarse en criterios clínicos y ser individualizada para cada paciente. Si el monitor fue indicado por un episodio de ALTE severo, deben transcurrir al menos 2 meses, contando desde el último episodio. Un paciente con antecedentes de apneas y medicado con xantinas, debe continuar monitorizado durante 1 mes luego de suspendida la medicación.
BIBLIOGRAFIA
Brooks J. G. Apparent life-threatening events and apnea of infancy. Clin. Perinat. 1992; 19:8809- 838.
Canani S. Wiebke J. Givan D. Temporal relationship between obstructive apnea and gastroesophageal reflux in infants. Pediatr. Pulmonol. 1997; 24:449. Abstract.
Cote A., Hum C., Brouillette R. T., Themens M. Frequency and timing of recurrent events in infants using home cardiorespiratory monitors. J Pediatr 1998; 132:783-789.
Davies A. E. M., Sandhu B. K. Diagnosis and treatment of gastro-oesophageal reflux. Arch Dis Child 1995; 73:82-86.
Ewer A. K., James M. E., Tobin J. M. Prone and left lateral positioning reduce gastro-oesophageal reflux in preterm infants. Arch Dis Fetal & Neonatal Ed 1999; 81:F201-F205.
Figueroa Turienzo J. M. Evento aparentemente amenazador para la vida. Medicina Infantil. 1996; 3:105114.
Hampton F. J., Mac Fadyen U. M., Simpson H. Reproducibility of 24 -hour esophageal pH studies in infants. Arch Dis Child 1990; 65:1249-54.
Hunt C., Corwin M., Lister G. et al. Longitudinal assessment of hemoglobin oxygen saturation in healthy infants during the first 6 months of age. J Pediatr 1999; 135:580-6.
Jeffery H. and Page M. Gastro-esophageal reflux in infants. SIDS Global Strategy Task Force: Development Physiology. Global Strategy task Force. Update report on Activities Since Washington DC (1996). Rouen, France 1998.
Kahn A: Conferencia "Back to the Future". Reducing the risks of SIDS. 16 th Annual Conference on Sleep Disorders in Infancy and Childhood - Palm Springs 1998.
Khoshoo V., Edell D., Clarke R. Effect of Cisapride on the QT interval in infants with gastroesophageal reflux. Pediatrics 2000; 105 (2).
Kneyber M. C., Brandenburg A. H., de Groot R. et al. Risk factors for respiratory syncycial virus associated apnoea. Eur J Pediatr 1998 Apr; 157(4): 4331-335.
Loughin G. M., Carroll J. Apparent life-threatening events. In Oskis Pediatrics. Principle and Practice 3ª. Ed., Baltimore, Lippincott Williams and Wilkins, 1999, Cap 103.
Lucey J. K. Comments on a sudden infant death article in another journal. Pediatrics 1999; 103:812.
Mahajan L., Wyllie R., Oliva L., Balsells F., Steffen, Kay M. Reproducibiluity of 24-hour intraesophageal pH monitoring in pediatric patients. Pediatrics 1998; 101:260-3.
Malloy M. H., Hoffman H. Home apnea monitoring and sudden infant death syndrome. Prev Med 1996; 25:645-649.
Mazzola M. L. Muerte súbita del lactante y episodios de aparente amenaza a la vida. Simposio Latinoamericano sobre el Síndrome de Muerte Súbita del Lactante. Buenos Aires 1999. Conferencia.
National Institutes of Health Consensus Development Conference on Infant Apnea and Home Monitoring Consensus statement. Pediatrics 1987; 72:292-299.
Oren J. Kelly D. Shannon D. Identification of a high-risk group for sudden infant death syndrome among infants who were resuscitated for sleep apnea. Pediatrics 1987; 292-299.
Orenstein S. Management of supraesophageal complications of gastroesophageal reflux disease in infants and children. Am J Med 2000; 108 (4ª): 139S-143S.
Peter C. S., Sprodowski N., Bohnhorst B., Jsilny, Poets C. F.. Gastroesophageal reflux and Apnea of Prematurity: Is there a relationship? Sixth SIDS International Conference. Auckland, New Zealand 2000. Abstract.
Rocca Rivarola M. Jenik A. Kenny P. Agosta G. Ruiz A. L. y Gianantonio C. Eventos de aparente amenaza a la vida. Experiencia de un enfoque pedíatrico interdisciplinario. Arch Arg Pediatr 1995; 93.: 85-91.
Schwartz P. J., Stramba-Badiale M., Segantini A. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med 1998; 338: 1709-1714.
Tirosh E. The relationship between gastroesophageal reflux (GER) and apnea of infancy. 5th International Conference. Rouen 1998. Abstract.
Tobin J. M., McCloud P., Cameron D. J. S. Posture and gastroesophageal reflux: a case for left lateral positioning. Arch. Dis. Child. 1997; 76:254-258.
Ward R. M., Lemons J. A., Molteni R. A. Cisapride: a survey of the frecuency of use and adverse events in premature newborns. Pediatrics 1999; 103: 469 - 472.
Weese-Mayer D. E., Brouilette R. T., Morrow A. S. et al. Assessing validity on infant monitor alarms with event recording. J Pediatrics 1989; 115: 702-708.
William S. M., Mitchell E. A., Stewart A. W. and Taylor B. J. Temperature and sudden infant death syndrome. Paediatr Perinat Epidemiol 1996 ; 10 (2):136-149.
5. DISPLASIA BRONCOPULMONAR
Gabriela Bauer
La displasia broncopulmonar (DBP) es hoy la enfermedad pulmonar crónica más frecuente del lactante y constituye la causa principal de morbilidad y mortalidad en los primeros años de vida de niños que fueron prematuros al nacer.
La incidencia de esta enfermedad guarda una estrecha relación con la sobrevida de recién nacidos (RN) con prematurez extrema y muy bajo peso ya que éstos constituyen el grupo mas vulnerable para contraer displasia broncopulmonar.
Una publicación realizada por la Universidad de Washington de una revisión multicéntrica de 3 años (jun 89/jul 91) muestra una incidencia de DBP de 87% en RN con peso de nacimiento entre 600 y 800gr (sobrevida: 79%); de36% en el grupo con peso entre 1000 y 1250gr y de 3.5% en el grupo con peso entre 1500 y 2000 gr (sobrevida: 97%). En los EE.UU. la prevalencia de DBP con una incidencia de prematurez del 15%, es de 1300 casos por año.
En nuestro medio carecemos de publicaciones estadísticas que comprendan varios centros de asistencia a Recién Nacidos y analicen cifras de incidencia de DBP y su relación con sobrevida y factores de riesgo.
Definición
Desde la descripción original de la displasia broncopulmonar hecha por W. H. Northway en 1967, se han utilizado diferentes criterios descriptivos lo que ha generado dificultades para comparar estudios epidemiológicos. Se propone la utilización de los criterios propuestos por el Bureau of Maternal and Child Health and Resources Development que fueron publicados en Pediatric Pulmonology, 1989.
1) Ventilación con presión positiva durante las 2 primeras semanas de vida con un mínimo de 3 días.
2) Signos clínicos de dificultad respiratoria (ej: taquipnea, aumento del esfuerzo respiratorio) persistentes mas allá de los 28 días de vida y/o 36 semanas postconcepcionales.
3) Requerimiento de oxígeno suplementario mas allá de los 28 días de vida y/o 36 semanas postconcepcionales para mantener una PaO2 por encima de 50 mmHg.
4) Cambios radiológicos con hallazgos anormales difusos característicos de DBP.
Etiopatogenia
La etiopatogenia de esta enfermedad puede representarse como un modelo de injuria y reparación en un pulmón inmaduro. La toxicidad por oxígeno y el barotrauma (estiramiento alveolar) juegan un rol principal entre los mecanismos de injuria y otros factores como el edema y el hiperflujo pulmonar,las infecciones respiratorias, la aspiración meconial, la desnutrición y los déficit vitamínicos actúan como coadyuvantes. El daño desencadenará una respuesta inflamatoria y si luego sucede una reparación anormal el recién nacido evolucionará con signos de enfermedad pulmonar crónica (EPC).
Los hallazgos anatomopatológicos encontrados en autopsias de pulmones de pacientes con DBP (presentes en grado variable y con áreas adyacentes de tejido sano) son: metaplasia del epitelio de la vía aérea superior, hipertrofia del músculo bronquial, destrucción de septum alveolares, extensas áreas saculares (detención en la septación), hipertrofia del músculo liso de los vasos acinares, fibrosis pulmonar.
Fisiopatología
Las consecuencias fisiopatológicas posibles (en distinto grado según el compromiso pulmonar crónico) en pacientes con DBP pueden ser: alteración del intercambio gaseoso (alt. de la V/Q), hipoxemia y en las formas más severas hipercapnia, aumento de la resistencia de la vía aérea, anormalidades del flujo traqueobronquial, flujo espiratorio limitado, reactividad de la vía aérea, atrapamiento aéreo, disminución de la compliance pulmonar, elevada presión intrapleural, aumento del trabajo respiratorio
Evolución clínica
En la evolución clínica de la displasia broncopulmonar pueden considerarse un período agudo en el que el paciente presenta labilidad del aspecto respiratorio y nutricional, requiriendo asistencia respiratoria (ARM) y aporte nutricional parenteral, un período de convalecencia en el que se encuentra con mayor estabilidad, en el que es posible plantear la salida de la ARM y podrá recibir alimentación enteral exclusivamente y el período de cronicidad donde el paciente requerirá o no oxigenoterapia suplementaria con distinto grado de compromiso respiratorio y en el cual se espera un crecimiento compensatorio. Es en esta última etapa donde será posible plantear el alta de la UCIN y comenzar el seguimiento ambulatorio.
SEGUIMIENTO
Aspectos a tener en cuenta
Algunos aspectos para ser tenidos en cuenta durante el seguimiento de pacientes con Displasia broncopulmonar son:
1. Aspecto respiratorio
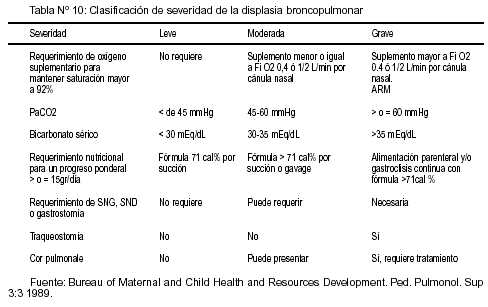
Los niños con DBP presentan distinto grado de severidad de la enfermedad. Se propone la utilización de una clasificación en estadíos leve, moderado y grave (ver cuadro) que considera el compromiso respiratorio (según el niño requiera oxigenoterapia y/o tenga EAB con hipercapnia); nutricional (según requerimiento calórico) y complejidad de cuidado requerido (serán graves los pacientes con severo compromiso pulmonar y/o traqueostomía y/o gastrostomía).
Si bien la DBP es causa de importante morbilidad respiratoria; el hecho de ocurrir en un período de la vida en que el pulmón se halla genéticamente programado para la mayor velocidad de crecimiento, posibilitará que el curso de la enfermedad sea favorable para la mayoría de los pacientes durante los tres primeros años. Es esperable que un paciente que fue dado de alta con severidad moderada de su DBP durante el primer año pase a estadío leve y uno en estadío leve evolucione a la ausencia de signos de enfermedad pulmonar.
Los niños con DBP pueden presentar episodios de hipoxemia durante la alimentación, el llanto, el ejercicio y el sueño En el primer año de vida aproximadamente el 50% de los niños con DBP requerirá reinternaciones por causa respiratoria. Las complicaciones que podrán presentar son:
— Sibilancias crónicas o recurrentes.
— Bronquitis recurrente o crónica.
— Infecciones respiratorias agudas.
— Atelectasias.
— Edema pulmonar.
— Hipertensión pulmonar.
— Estenosis u otras complicaciones de la vía aérea superior.
— Traqueomalacia.
Publicaciones sobre secuelas pulmonares en adolescentes y adultos jóvenes con antecedentes de DBP en su infancia concluyen que la mayoría de éstos presentó algún grado de disfunción (obstrucción y/o hiperreactividad de la vía aérea, hiperinflación etc.)
2. Aspecto nutricional
Los niños con DBP que son dados de alta de las unidades neonatales suelen presentar varios factores de malnutrición (peso y talla anormal, osteopenia, masa muscular pobre etc.).
Los pacientes con DBP moderada a severa pueden tener un aumento del metabolismo basal por aumento del consumo de oxígeno y del esfuerzo respiratorio por una mecánica pulmonar anormal.Se han publicado déficits de pre albúmina que sumaría un factor de tipo proteico a la desnutrición calórica que pueden presentar.
Estos pacientes podrán presentar dificultades para lograr un adecuado aporte calórico: patrones de succión anormales dificultad respiratoria que interfiere con la alimentación (taquipnea, tos), alteraciones del paladar, trastornos de deglución, reflujo gastroesofágico, limitación iatrogénica de líquidos.
Los niños que reciben diuréticos en forma crónica pueden presentar trastornos metabólicos y/o electrolíticos: alcalosis metabólica, hipocloremia, hipopotasemia, hiperglucemia, hipocalcemia con hipercalciuria.
En estudios a largo plazo niños de 10 años con antecedentes de DBP presentaron valores de talla y peso en un 10% inferiores a los de controles.
3. Problemas del desarrollo
Además de la enfermedad pulmonar los niños pueden tener problemas neurológicos o del desarrollo relacionados con la prematurez, hemorragia intracraneana, asfixia perinatal.
Si bien las opiniones sobre este tema permanecen controvertidas existen evidencias de que los pacientes con DBP presentarían mayor riesgo de retraso madurativo independientemente de otros eventos perinatales asociados.
Algunos de los déficit motores observados en los 2 primeros años de vida pueden ser transitorios y relacionarse con la enfermedad crónica (limitación de la actividad, reinternaciones prolongadas, desnutrición). Trastornos cognitivos tales como alteraciones perceptivo-motoras, no pueden ser valoradas hasta la edad escolar.
4. Aspecto vincular y social
Los niños con displasia broncopulmonar moderada o severa pueden manifestar displacer, irritabilidad, poco interés, presentar dificultad para alimentarse o para ser consolados, retraso en la sociabilización, esto podrá ocasionar dificultades vinculares.
La evolución de este complejo y crónico síndrome demanda de las familias máximas necesidades de adaptación.
El fuerte impacto que produce asumir tratamientos como la oxigenoterapia prolongada y frecuentes reinternaciones implica un elevado costo emocional y económico para el medio del niño enfermo.
5. Otros aspectos
Los pacientes con DBP pueden padecer otras complicaciones - Cardiovasculares: hipertensión arterial sistémica y/o pulmonar en distintos grados.
— Digestivas (reflujo gastroesofágico).
— Apneas-riesgo aumentado de muerte súbita (controvertido).
OBJETIVOS DEL SEGUIMIENTO
1. Minimizar el daño pulmonar
a -Prevención y tratamiento de infecciones respiratorias
Calendario de vacunas actualizado para la edad cronológica. Vacunas especiales: antigripal en niños mayores de 6 meses: 2 dosis el 1er año (0,25ml c/u) luego una dosis anual en el mes de marzo, mientras tenga signos de EPC. En lactantes menores vacunar a padres o cuidadores.
Vacuna antineumocóccica: 1 dosis a niños mayores de 2 años que persistan con signos de EPC a esa edad o vacuna conjugada heptavalente antes de los dos años (Ver Capítulo 3 en Inmunzaciones).
Educación a la familia en medidas de aislamiento de contactos, evitar hacinamiento y guarderías, pautas de detección temprana de signos de infección respiratoria aguda (IRA) y consulta precoz.
Si el niño tiene signos de IRA control médico, en especial evaluar requerimiento de oxigenoterapia por oximetría de pulso, de no ser posible y el paciente presenta signos de dificultad respiratoria moderada a severa se recomienda internación para mejor control evolutivo y oxigenoterapia además de otros tratamientos.
b- Disminuir la toxicidad ambiental (exposición a humos).
Se indicará en todos los casos no fumar en los ambientes donde el niño permanezca, no usar braseros ni estufas a kerosén y en el caso de calefacción con estufa de cuarzo humedificar el ambiente.
c - Tratamiento adecuado de la obstrucción bronquial y del edema pulmonar
Una adecuada indicación de tratamientos puede basarse en el reconocimiento de los signos y síntomas que el niño con DBP presente en lo que se considere su "situación basal", para así poder hacer un correcto diagnóstico de desmejorías y evaluar causas y manejo de las mismas.
Si el paciente presenta signos que expresan obstrucción al flujo de la vía aérea intentar definir si son permanentes o recurrentes.
En términos generales se recomienda el uso de series de broncodilatadores inhalados 2 agonistas a dosis adecuadas al peso, evitando el uso crónico de los mismos dados los efectos adversos que pueden presentarse y el desarrollo de tolerancia. En crisis de obstrucción bronquial de mayor intensidad asociar tratamiento con corticoides orales o sistémicos durante el menor tiempo posible necesario.
En niños con reiterados episodios de sibilancias puede plantearse el uso de medicación preventiva con Cromoglicato disódico inhalado y evaluar beneficio.
Algunos pacientes evolucionan con bronquitis crónica o recurrente, pudiendo beneficiarse con kinesioterapia respiratoria.
Si el paciente tiene signos de edema pulmonar se recomienda efectuar un correcto balance de ingreso y egreso. En primer término restringir ingesta de líquidos y evaluar respuesta. Si persiste con signos asociar diurético (fursemida una o dos dosis diarias de 1 mg/kg) luego de 2 a 3 días evaluar beneficio y necesidad por ionograma de indicar ClK. Si hay buena respuesta pasar a tratamiento en días alternos una dosis diaria y suspender en cuanto el niño mejore.
2) Promover el crecimiento de nuevo tejido pulmonar.
— Estricto control nutricional: Intentar cálculo correcto de las necesidades nutricionales y evaluar que el paciente pueda recibirlas. Se recomienda el uso de recordatorios por escrito, observar al niño en las visitas mientras lo alimentan, interrogar a la madre sobre problemas que suelen asociarse con la alimentación. En algunos niños muy sintomáticos puede ser necesario el uso prolongado de SNG para alimentación, en forma exclusiva o parcial.
Se estima que durante el primer año los niños con DBP moderada a leve pueden requerir aportes de entre 140 y 160 cal/kg/día. Si es necesario controlar la ingesta de líquidos pueden utilizarse los sucedáneos de la leche materna preparados al 20% y enriquecidos con cereales. Los suplementos de minerales y hierro se indicarán como lo recomendado para prematuros. (Ver Capítulo sobre Alimentación).
3) Mantener una óptima saturación en todo momento
Saturometrías seriadas, considerándose adecuados valores mayores o iguales a 92%. Se recomienda el uso de oxímetro de pulso para control de saturación en los controles médicos de los niños con DBP mientras continúen con signos de enfermedad pulmonar. Replantear en un paciente que no evoluciona favorablemente la necesidad de oxigenoterapia crónica, efectuando monitoreo prolongado en distintas situaciones (vigilia, comiendo, llorando, durmiendo).
Durante y luego de padecer una IRA un niño puede aumentar los requerimientos de oxígeno suplementario, o requerirlo si no lo tenía previamente.
Previo a indicar suspensión del tratamiento efectuar una evaluación cardiológica, el niño no debe tener signos de Hipertensión Pulmonar (HP). Es recomendable el traslado del paciente en ambulancia o móvil sanitario a los controles médicos, durante el tiempo que requiera oxigenoterapia domiciliaria. (Ver Anexo Nº 7).
BIBLIOGRAFIA
Avery G. B.,Tooley W. H., Keller J. B. et al: Is chronic lung disease in low birth weigth infants preventable? A survey of 8 centers. Pediatrics 1987; 79:26-30.
Bureau of Maternal and Child Health and Resources Development. Ped. Pulmonol. Sup 3: 3 1989.
Farrell P., Fascone J. Bronchopulmonary dysplasia in 1990’s: A review for the Pediatrician. Curr Probl Pediatr, april 1997.
González Pena H., Grenoville M. Oxigenoterapia domiciliaria. Medicina Infantil, 5(4): 273-276, 1998.
Hazinsky T. A. Bronchopulmonary dysplasia in disorders of respiratory tract in children. Section II: Respiratory disorders in the newborn. 6ª Ed. Philadelphia, W. B. Saunders Co., 1998.
Northway W. H., Rosan R. C., Porter D. Y.: Pulmonary disease following respirator therapy of HMD. N Engl J Med 1967; 276:357-368.
6. ANEMIA EN EL PREMATURO
Patricia Climent
Todos los niños experimentan un descenso progresivo en la hemoglobina, hematocrito y número de glóbulos rojos como consecuencia de la disminución del estímulo eritropoyético que genera el aumento de oxigenación tisular que se produce al nacer. En los niños prematuros los cambios se inician más precozmente y son más intensos.
Etapas de la eritropoyesis posnatal
Anemia temprana o fisiológica: Es fisiológica ya que se conservan los mecanismos adaptativos que preservan la disponibilidad de oxígeno para los tejidos. Está determinada por la disminución en la producción de eritropoyetina y en la vida media de los glóbulos rojos. Se extiende durante 4 a 8 semanas.
Después de la primer semana de vida la médula presenta una brusca interrupción en su funcionamiento. La eritropoyesis prácticamente se interrumpe con el nacimiento y no comienza hasta después del período neonatal • Disminuyen los precursores eritroides.
• Disminuye el recuento de reticulocitos.
• Disminuye el nivel de hemoglobina.
• Aumenta la ferritina.
Los depósitos de hierro aumentan o se mantienen estables si no hay pérdida de sangre.
La magnitud de la caída de todos estos elementos, la rapidez con que se llega al nivel más bajo y la persistencia en valores mínimos están relacionados al peso de nacimiento, la edad de gestación y el sexo del niño.
A las 32 semanas de gestación los varones han alcanzado valores de hemoglobina (16,3gr) iguales a los del recién nacido de término. En las niñas, este valor máximo no se alcanza hasta haber completado las 39 semanas. Por lo tanto, al nacer prematuramente, el sexo femenino tiene valores más bajos que el masculino, dependiendo de su EG.
La oxigenación tisular depende de la concentración de hemoglobina, la afinidad de la hemoglobina por el oxígeno y la interacción entre hemoglobina y 2,3 difosfoglicerato.
Frente a descensos similares en los niveles de hemoglobina, los niños más inmaduros tienen menor capacidad de producir eritropoyetina que niños mayores o adultos. Esto sumado a la mayor afinidad de la hemoglobina fetal por el oxígeno, determina que la liberación de oxígeno hacia los tejidos en el prematuro anémico esté muy restringida y pueda ocasionar síntomas.
A pesar de los mecanismos compensadores fisiológicos (aumento de la eritropoyetina plasmática, desviación hacia la derecha de la curva de disociación de la oxihemoglobina y aumento de la extracción de oxígeno), por la disminución progresiva de la concentración de hemoglobina, algunos niños de bajo peso tendrán síntomas de anemia. Los síntomas de "disponibilidad de oxígeno" inadecuada se manifiestan por anorexia, taquicardia, taquipnea, letargo, episodios apneicos, rechazo o fatiga por la alimentación.
El uso de eritropoyetina recombinante ha hecho disminuir la necesidad de transfusiones.
Los niños que han sido repetidamente transfundidos tendrán predominancia de hemoglobina A, que libera oxígeno más fácilmente que la hemoglobina F, determinando un estímulo eritropoyético menor expresado por menor número de reticulocitos para un nivel determinado de hemoglobina. Estos niños toleran niveles de hemoglobina más bajos que aquellos que no han recibido transfusiones. Ellos deberán estar más anémicos antes que se estimule la producción de eritropoyetina. Por lo tanto los valores de hemoglobina o hematocrito con los cuales se podrá producir hipoxia tisular son diferentes para los recién nacidos que han recibido transfusiones múltiples en comparación con aquellos que no han sido transfundidos.
La liberación de eritropoyetina se iniciará con niveles más bajos de hematocrito en los niños politransfundidos. El efecto depresor de la eritropoyesis provocada por la transfusión, en la etapa de cuidados intensivos, puede mantenerse cuando ésta ha cesado.
Tener en cuenta que cuando no ha habido reposición de las pérdidas, las reservas de hierro serán menores y habrá más riesgo de deficiencia.
La duración de las etapas está señalada por las líneas punteadas. I. Anemia fisiológica o temprana. II. Fase de recuperación. III. Anemia tardía.
2. Fase de recuperación:
Cuando los niveles de hemoglobina generan hipoxia tisular se reanuda la actividad eritropoyética de la médula ósea. Esto sucede entre el segundo y tercer mes de vida. Al quinto mes de vida el niño prematuro habrá alcanzado niveles de hemoglobina semejantes a los del recién nacido de término.
• Aumentan los precursores eritroides en medula ósea.
• Aumenta la eritropoyetina.
• Aumentan los reticulocitos.
Se mantiene el nivel de hemoglobina (la hemoglobina comienza a aumentar una semana después de iniciado el aumento de reticulocitos).
• Disminuye la ferritina.
Cuando la anemia es lo suficientemente severa se inicia la liberación de eritropoyetina. Hay una semana de demora entre el momento en que se eleva la eritropoyetina y el momento en que aparecen reticulocitos en circulación. Hay otra semana de demora antes que la hemoglobina y el hematocrito comiencen a elevarse. Cuando la médula se ha activado se produce hemoglobina F aunque los niños hayan sido transfundidos con hemoglobina A. Recién después de las 34 semanas la producción de hemoglobina F comienza a disminuir y va aumentando la de A.
Si el niño ha iniciado durante este tiempo su crecimiento compensatorio, el aumento de volumen plasmático puede ser más rápido que el de la masa globular y por lo tanto los valores de hemoglobina se mantendrán estables a pesar de existir eritropoyesis activa.
La suplementación con hierro no modifica la duración de estas etapas aunque el prematuro comienza a depender más precozmente del aporte de hierro por ser menores sus reservas.
3. Anemia tardía o nutricional
Más allá del 3er o 4to mes la anemia será dependiente del aporte de hierro.
Las demandas para la síntesis de hemoglobina sobrepasan las reservas y cuando el peso se duplica hay un agotamiento de las mismas. Si no se repusieron las pérdidas por extracciones o si atraviesa el período de crecimiento compensatorio, el déficit será aún mayor.
Varias de las siguientes situaciones pueden agravar el cuadro:
• Transfusiones feto-fetales (embarazos gemelares).
• Exsanguinotransfusión en período neonatal.
• Antecedente de hemorragias o extracciones reiteradas.
Puede comenzarse la administración de hierro cuando reciba alimentación enteral desde la segunda semana o al duplicar el peso de nacimiento.
|
. |
RN PRETERMINO |
RN TERMINO |
|
Valor inferior de Hb g % |
7-9 |
10-11 |
|
Alcanzado a Respuesta Reticulocitaria |
4ª a 8ª semana más precoz |
6ª a 12ª semana más tardía |
Recomendaciones para la administración de Fe en pretérminos:
• desde duplicación del peso de nacimiento.
• desde 2 semanas (controvertido).
• niños poli-transfundidos pueden recibir el mismo régimen.
• dosis 2 mg/kg/día (no exceder de 15 mg/día) hasta 12 – 15 meses de vida.
• 1000 a 2000 g de PN: 2 mg/kg/día, < de 1000 g de PN: 4mg /kg /día.
OTRAS ANEMIAS NUTRICIONALES:
Anemia por déficit de ácido Fólico:
Luego del nacimiento los niveles séricos disminuyen en todos los recién nacidos. Mientras que la leche humana y las fórmulas cubren los requerimientos del niño de término, pueden resultar insuficientes para el pretérmino en crecimiento rápido por lo que se recomienda la administración de 50 microgramos diarios durante los primeros 8 meses. Este aporte está cubierto con la ingestión de medio litro de cualquier fórmula infantil. La deficiencia de ácido fólico puede ocasionar anemia megalobástica, que aparece generalmente durante los tres primeros meses; más aún si el prematuro ha padecido infecciones recurrentes o diarrea.
Déficit de Vitamina B 12:
Los niños que fueron sometidos a resecciones intestinales del íleon terminal tienen riesgo aumentado de padecer deficiencia de vitamina B 12.
La aparición de macrocitosis, hipersegmentación de los neutrófilos, indicará la necesidad de administrarla en forma parenteral a intervalos regulares.
Deficiencia de Vitamina E:
Los prematuros presentan un déficit de absorción de vitamina E que persiste hasta los 2 a 3 meses, cuando no se administraron los suplementos adecuados o cuando exista falta de equilibrio entre aportes de Fe, ácidos grasos poliinsaturados y vitamina E.
Entre la 4ta y 6ta semana puede aparecer anemia hemolítica normocrómica por aumento de la fragilidad del eritrocito.
Luego del alta el intestino debería haber logrado capacidad de absorber grasas y las necesidades estarían cubiertas por la dieta, sin embargo algunos niños que no han alcanzado los 2500 gramos, pueden requerir suplementos hasta un mes después.
Deficiencia de Cobre
Puede presentarse en niños con problemas enterales o que requirieron nutrición parenteral prolongada.
Puede ser causa de anemia microcítica e hipocrómica –sospecharla cuando el suplemento (80 microgramos diarios) no ha sido el adecuado– especialmente si el pretérmino es BPEG ya que tiene una disminución en la síntesis de ceruloplasmina hepática. Se presentan entre el segundo y tercer mes de vida y se acompaña de niveles de cupremia inferiores a 40 microgramos.
RECOMENDACIONES PARA EL SEGUIMIENTO DE LA ANEMIA EN EL RECIEN NACIDO PRETERMINO:
• Durante la primer semana de vida mantener la Hb en 13 g en los niños de < de 1.500g. En presencia de SDR se aconseja niveles más altos (16 g).
• Registrar extracciones efectuadas para estudios de laboratorio (recordar que el volumen sanguíneo es de 80 a 85 ml/kg).
• Controlar los valores de Hb durante la internación en la 1ª semana y luego una vez por semana.
• Suplementar con vitamina E, 5 a 50 u por día en los menores de 34 semanas de EG hasta el término. Mantener una relación adecuada vitamina E/ácidos grasos polinsaturados. En la leche materna y en las fórmulas maternizadas esta relación es adecuada.
• Recomendación para administración de Fe en prematuros. Si la respuesta es insatisfactoria, solicitar: Hb, frotis, índices hematimétricos, reticulocitos, y eventualmente consulta hematología.
Luego del alta:
Tener en cuenta que los valores fisiológicos varían de acuerdo al peso de nacimiento, edad gestacional y edad posnatal.
Si el hematocrito al alta es del 25% o menor realizar control clínico en 72 hs y alertar a los padres sobre signos de alarma.
Solicitar hemograma con recuento reticulocitario y frotis.
Si el hematocrito se mantiene estable revisar aportes y continuar con controles clínicos cada tres días hasta lograr valores iguales a la media.
Si el hematocrito es mayor del 25% repetir control a los 15 días, si persiste igual tendencia efectuar nuevos controles a los 3, 6 y 12 meses.
Si al alta el hematocrito está entre 25 y 30 % controlar entre una y dos semanas.
Si se mantiene estable continuar controles a los 3, 6 y 12 meses.
Anemia:
Cuándo preocuparse:
• Cuando el niño presente síntomas clínicos, taquicardia, taquipnea, dificultad en alimentarse, disminución de la actividad, escasa ganancia de peso.
• Si el nivel de hemoglobina no comenzó a aumentar a los 4 meses de EG Corregida.
• Si continua el nivel de disminución de hemoglobina después de los 4 meses a pesar de Fe, ácido fólico, Vit. E y terapia con Cu.
• Si el niño presenta alguna otra secuela: EPC, ostomía luego de cirugía intestinal o enfermedad residual renal.
• Si los índices hematimétricos son anormales.
Transfusión:
La indicación de transfusiones en niños de bajo peso al nacer debe basarse en criterios clínicos objetivos y no en niveles de hemoglobina establecidos arbitrariamente.
Muchos niños pretérmino sobre todo los que han recibido múltiples transfusiones están saludables con niveles de hemoglobina de 6 a 7 g/dl, otros con valores mayores requieren tratamiento.
Al considerar la interpretación de la concentración de hemoglobina y la necesidad de posibles formas alternativas de tratamiento deben incluirse los siguientes factores: EG y edad posnatal, estado clínico (frecuencia cardíaca y respiratoria, saturación arterial de oxígeno, presencia de letargo, fatigabilidad fácil al alimentarse, antecedentes de mal aumento de peso), el nivel inicial de hemoglobina, el volumen de sangre extraída para estudios diagnósticos y no repuesta y los antecedentes de transfusiones previas. Si existe enfermedad respiratoria la decisión de transfundir será más precoz.
Considerar riesgos potenciales de las transfusiones: transmisión de hepatitis B, C, no A no B, HIV, citomegalovirus, sobrecargas de volumen, reacciones inmunitarias.
El volumen a transfundir será el suficiente para lograr mejoría de los síntomas. Generalmente es suficiente elevar el hematocrito hasta 30%.
Hemoglobina (g/100 ml) (valores medios)
|
EDAD POSTNATAL |
3 días |
6 semanas |
8 semanas |
10 semanas |
|
PN Menor de 1500 gramos EG 28-32 semanas |
17,5 |
8,5 |
8,5 |
9 |
|
Hematocrito (%) |
54 |
25 |
25 |
28 |
|
Recuento de reticulocitos (%) |
8 |
11 |
8,5 |
7 |
BIBLIOGRAFIA
Burman, Morris A. F. Cord hemoglobin in low birth weight infants. Arch Dis Child, 1972; 49:382.
Jones D., Gleason C., Lipstein S. Postnatal anemia in hospital care of the recovering NICU infant. Baltimore, Williams and Wilkins, 1991.
Koerper M. Anemia in the ICN graduate. In Ballard. Pediatric care of the ICN Graduate. Philadelphia, Saunders, 1988.
Oski F., Naiman N. Problemas hematológicos del recién nacido. 3ª Ed. Buenos Aires, Panamericana, 1984.
Phibbs R., Sola A. Anemia en el recién nacido pretérmino en Sola A, Urman J. Cuidados Intensivos Neonatales, Buenos Aires, Interamericana, 1987.
Sabio H. Anemia in the High Risk Infant. Clinics in Perinatology. 1984; 11(1): 59-70.