ADMINISTRACIÓN
NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGÍA MÉDICA
Disposición 107/2024
DI-2024-107-APN-ANMAT#MS
Ciudad de Buenos Aires, 06/01/2024
VISTO el EX-2023-102515291-APN-DGA#ANMAT, las Disposiciones ANMAT Nº
3185/99 con sus complementarias y modificatorias, 5040/06 y sus
modificatorias, 4133/12, 1918/13, 6677/10 con sus complementarias y
modificatorias, 758/09, 4132/12, 9944/19, 3113/10, 9708/19, 9465/22,
5640/22, y
CONSIDERANDO:
Que por la Disposición ANMAT Nº3185/99 se aprobaron recomendaciones
para la realización de estudios de bioequivalencia de medicamentos de
riesgo sanitario significativo y se estableció un cronograma de
implementación gradual en consideración a los antecedentes
internacionales en la materia.
Que en la aludida Disposición se establecen los métodos idóneos para
demostrar equivalencia In Vivo y equivalencia In Vitro.
Que mediante disposiciones posteriores, la ANMAT ha ido incorporando
ingredientes farmacéuticos activos a la obligatoriedad de demostración
de estudios de bioequivalencia.
Que el grado de desarrollo alcanzado actualmente por el sistema
fiscalizador de nuestro país incluye el diseño y presentación de
protocolos de investigación en farmacología clínica, cuyos requisitos
se encuentran establecidos por la Disposición ANMAT N°6677/10, que
aprueba el Régimen de Buenas Prácticas Clínicas para Estudios en
Farmacología Clínica.
Que por la Disposición ANMAT Nº5040/06 se aprobó el Régimen de Buenas
Práctica para la realización de Estudios de Biodisponibilidad y
Bioequivalencia.
Que por la Disposición ANMAT Nº 4133/12 se estableció que solo se
deberán presentar resultados de estudios de bioequivalencia cuando los
mismos se hallen comprendidos dentro del intervalo de confianza del 90%
de 0,80-1,25 tanto para Concentración Máxima como para Área Bajo la
Curva.
Que por la Disposición ANMAT N° 1918/13 se establecieron los criterios
de selección de una especialidad medicinal como producto de referencia
para estudios de Bioequivalencia y Equivalencia in vitro.
Que por la Disposición ANMAT N° 9944/19 se establecieron los requisitos
y condiciones que deben cumplir los Centros Clínicos para ser
autorizados a realizar Estudios de Farmacología Clínica de
Bioequivalencia.
Que por la Disposición ANMAT N° 758/09 se adoptaron los Criterios de
Bioexencion de Estudios de Bioequivalencia basados en la Clasificación
Biofarmacéutica para las formas farmacéuticas sólidas orales de
liberación inmediata, con alcance para algunos de los principios
activos establecidos en la Disposición ANMAT N° 3185/99 como de riesgo
intermedio y los que en un futuro se vayan incorporando, así como las
exigencias que se deberán cumplir para demostración de Bioexención.
Que por la Disposición ANMAT N° 4132/12 se incorporó a la exigencia de
demostración de bioequivalencia, establecida en la Disposición ANMAT N°
3185/99, a todas las concentraciones de una especialidad medicinal de
forma farmacéutica sólida oral que contenga alguno de los IFAs con
requerimiento de bioequivalencia,
Que en el mercado local existen especialidades medicinales que
contienen IFAs que pueden ser tomados como referencia para la
realización de estos estudios.
Que en tal sentido el Servicio de Bioequivalencia del Departamento de
Ensayos Clínicos de la Dirección de Investigación Clínica y Gestión del
Registro de Medicamentos, se expidió en el
IF-2023-134567405-APN-DERM#ANMAT aconsejando la inclusión de nuevos
IFAs del grupo terapéutico antiepilépticos con indicación en el
tratamiento y prevención del síndrome convulsivo en la epilepsia a la
exigencia de realización de estudios de Bioequivalencia y/o
Biodisponibilidad.
Que en particular informó que los fármacos antiepilépticos utilizados
en la prevención y tratamiento del síndrome convulsivo en la epilepsia
constituyen un grupo terapéutico de uso crítico y de dosis crítica por
su indicación donde se debe asegurar la calidad de los mismos.
Que agregó que una inadecuada biodisponibilidad de los mismos, estando
por fuera del rango terapéutico puede ocasionar falta de eficacia con
la subsiguiente aparición de síndrome convulsivo así como también la
aparición de eventos adversos o toxicidad de los mismos.
Que por Disposición ANMAT N° 3185/99 se incorporó a los IFAs,
Carbamazepina, Fenitoína sódica, Valproato, Oxcarbazepina, y
Etosuximida, por Disposición ANMAT N° 9708/19 se incorporó al IFA
Pregabalina, por Disposición ANMAT N° 3113/10 se incorporó a los IFAs
Lamotrigina, y Topiramato, y por Disposición ANMAT N° 9465/22 se
incorporó al IFA Eslicarbazepina.
Que en consecuencia la aludida área técnica aconseja la inclusión de
los IFAs del grupo terapéutico antiepilépticos, con indicación en la
prevención o tratamiento del síndrome convulsivo, que se encuentren
registrados a la fecha, o los que en un futuro se inscriban en el
Registro de Especialidades Medicinales (REM), además de los que se
encuentran comprendidos dentro del listado de exigencia de estudios de
bioequivalencia in vivo o in vitro según corresponda con anterioridad.
Que asimismo en el Anexo I de la presente disposición se brinda
información sobre los Productos de Referencia a ser utilizados para los
IFAs ya registrados en el REM, que no se encuentran a la fecha en un
listado de exigencia de estudios de bioequivalencia in vivo o in vitro.
Que el Instituto Nacional de Medicamentos y la Dirección de Asuntos
Jurídicos han tomado la intervención de su competencia.
Que se actúa en virtud de las facultades conferidas por el Decreto
Nº1490/92 y sus modificatorios.
Por ello,
LA ADMINISTRADORA NACIONAL DE LA ADMINISTRACIÓN NACIONAL DE
MEDICAMENTOS, ALIMENTOS Y TECNOLOGÍA MÉDICA
DISPONE:
ARTÍCULO 1º.- Incorpóranse a la exigencia de realización de estudios de
Bioequivalencia establecida por la Disposición ANMAT Nº 3185/99, a los
Ingredientes Farmacéuticos Activos (IFAs) de forma farmacéutica sólida
oral que tengan como indicación la prevención o tratamiento del
síndrome convulsivo en el contexto de la epilepsia y pertenezcan al
grupo terapéutico de antiepilépticos, que se encuentren registrados a
la fecha o los que en un futuro se inscriban e incorporen en el REM,
además de los que se encuentran comprendidos en un listado de exigencia
de estudios de bioequivalencia in vivo o in vitro con anterioridad.
ARTICULO 2°.- Establécese que los productos de referencia a ser
utilizados para los IFAs registrados y que no se encuentren
comprendidos en un listado de exigencia de estudios de bioequivalencia
con anterioridad serán los que se mencionan en el Anexo que como
IF-2023-142995707-APN-DERM#ANMAT forma parte de la presente disposición.
ARTICULO 3°.- Los laboratorios titulares de especialidades medicinales
que tengan registrado o en trámite de registro algún IFA alcanzado por
el Artículo 1° de la presente disposición deberán presentar a partir de
la publicación de la presente disposición en el boletín oficial, en el
término de 180 días corridos ante esta Administración el protocolo de
estudio clínico de bioequivalencia de acuerdo a la normativa vigente,
Disposición ANMAT N° 5640/22, o la que en un futuro la modifique o
reemplace. Una vez obtenida la disposición autorizante del protocolo
por parte de esta Administración tendrá un plazo de 180 días corridos
para la presentación de los resultados del mismo.
ARTÍCULO 4º.- Para los laboratorios titulares de especialidades
medicinales con bioequivalencia demostrada para una de sus
concentraciones y que posean más de una concentración comercializada,
deberán cumplimentar las Disposiciones ANMAT Nº 758/09 y N° 4132/12, o
las que en un futuro las complementen o reemplacen, presentando los
protocolos de solicitud de bioexención para todas las concentraciones,
según la Disposición ANMAT Nº 5068/19, o la que en un futuro la
complemente o reemplace.
ARTÍCULO 5°.- La presente disposición entrará en vigencia a partir del
día siguiente al de su publicación en el Boletín Oficial.
ARTÍCULO 6°.- Regístrese. Dése a la Dirección Nacional del Registro
Oficial para su publicación. Dése a la Dirección de Gestión de
Información Técnica, al Instituto Nacional de Medicamentos, a la
Dirección de Investigación Clínica y Gestión del Registro de
Medicamentos y a la Dirección de Relaciones Institucionales.
Comuníquese a las Cámaras de la industria farmacéutica CILFA, CAEME,
COOPERALA, CAPGEN y CAPEMVEL, SAFyBI. Cumplido archívese.
Nelida Agustina Bisio
NOTA: El/los Anexo/s que integra/n este(a) Disposición se publican en
la edición web del BORA -www.boletinoficial.gob.ar-
e. 09/01/2024 N° 896/24 v. 09/01/2024
(Nota
Infoleg:
Los anexos referenciados en la presente norma han sido extraídos de la
edición web de Boletín Oficial)

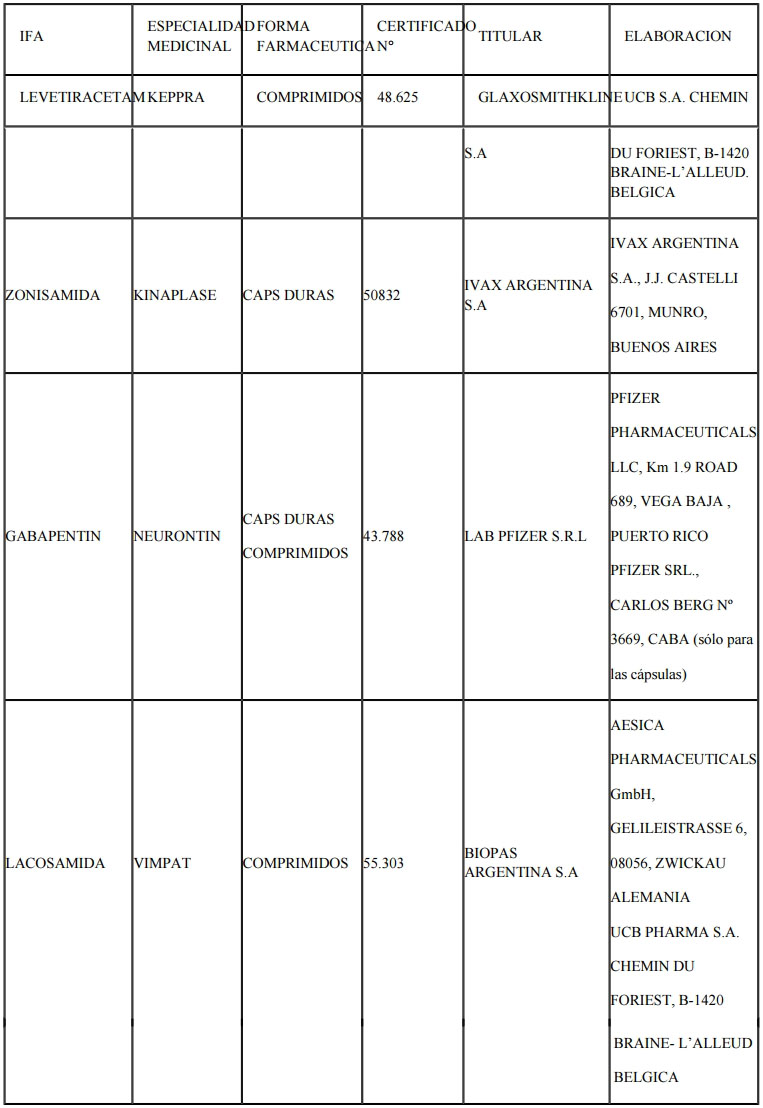
ANEXO
I
INGREDIENTES FARMACÉUTICOS ACTIVOS (IFAs) DEL GRUPO TERAPÉUTICO
ANTIEPILEPTICOS YA REGISTRADOS EN EL R.E.M SIN EXIGENCIA DE ESTUDIOS
CLINICOS DE BIOEQUIVALENCIA A LA FECHA QUE PASAN A FORMAR PARTE DEL
LISTADO DE EXIGENCIA DE DICHOS ESTUDIOS A PARTIR DE LA ENTRADA EN
VIGENCIA DE LA PRESENTE DISPOSICION:
LEVETIRACETAM
ZONISAMIDA
GABAPENTIN
LACOSAMIDA
PRODUCTOS DE REFERENCIA A SER UTILIZADOS PARA LOS IFAs ANTIEPILEPTICOS
COMERCIALIZADOS SIN EXIGENCIA DE ESTUDIOS DE BIOEQUIVALENCIA A LA FECHA
QUE PASAN A FORMAR PARTE DEL LISTADO DE EXIGENCIA A PARTIR DE LA
ENTRADA EN VIGENCIA DE LA PRESENTE DISPOSICION: