MERCOSUR/GMC/RES. N° 20/17
PROCEDIMIENTOS
COMUNES PARA LAS INSPECCIONES A LOS FABRICANTES DE PRODUCTOS MÉDICOS Y
PRODUCTOS PARA DIAGNÓSTICO DE USO IN VITRO EN LOS ESTADOS PARTES
(DEROGACIÓN DE LA RES. GMC N° 23/15)
VISTO: El Tratado de Asunción,
el Protocolo de Ouro Preto y la Resolución N° 23/15 del Grupo Mercado
Común.
CONSIDERANDO:
Que se deben actualizar los procedimientos comunes para la realización
de inspecciones sanitarias a los fabricantes de Productos Médicos y
Productos para Diagnóstico de Uso In Vitro, de acuerdo con la
experiencia adquirida en el desarrollo de acciones conjuntas en el
ámbito del MERCOSUR.
Que la actualización de los requisitos de Buenas Prácticas de
Fabricación de Productos Médicos y Productos para Diagnóstico de Uso In
Vitro requiere que el sistema de las inspecciones se base en el
análisis de riesgos.
Que es necesario adoptar criterios comunes para la toma de decisiones
de acuerdo a los resultados de la inspección.
EL
GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar los "Procedimientos comunes para las inspecciones a
los fabricantes de productos médicos y productos para diagnóstico de
uso In Vitro en los Estados Partes”, que constan como Anexo y forman
parte de la presente Resolución.
Art. 2 - Los Estados Partes indicarán en el ámbito del Subgrupo de
Trabajo N° 11 “Salud” (SGT N° 11) los organismos nacionales competentes
para la implementación de la presente Resolución.
Art. 3 - Derogar la Resolución GMC N° 23/15.
Art. 4 - Esta Resolución deberá ser incorporada al ordenamiento
jurídico de los Estados Partes antes del 31/XII/2017.
XLVIII
GMC EXT. - Mendoza, 19/VII/17.
ANEXO
PROCEDIMIENTOS COMUNES PARA LAS
INSPECCIONES A LOS FABRICANTES DE PRODUCTOS MÉDICOS Y PRODUCTOS PARA
DIAGNÓSTICO DE USO IN VITRO EN
LOS ESTADOS PARTES
1. OBJETIVO
Establecer procedimientos para la realización de inspecciones en los
establecimientos que fabrican productos médicos y productos para
diagnóstico de uso in vitro, así como criterios comunes para la toma de
decisiones de acuerdo a los resultados de la inspección.
2. ÁMBITO DE APLICACIÓN
Estos procedimientos se aplican a las inspecciones intrazona realizadas
por los Estados Partes en establecimientos fabricantes de productos
médicos y productos para diagnóstico de uso in vitro, comercializados
entre los Estados Partes, en las siguientes situaciones:
a) emisión del Certificado de Buenas
Prácticas de Fabricación;
b) verificación de la rutina del cumplimiento de las Buenas Prácticas
de Fabricación;
c) verificación del cumplimiento de las adecuaciones requeridas en la
inspección previa;
d) investigación
de los informes de eventos adversos, reclamos y reportes de
irregularidades.
3. TOMA DE DECISIONES EN RELACIÓN AL
CUMPLIMIENTO DE BUENAS PRÁCTICAS DE FABRICACIÓN (BPF)
El otorgamiento del Certificado de Buenas Prácticas de Fabricación, se
fundamentará en los resultados de la evaluación del cumplimiento de los
requisitos de BPF, teniendo en cuenta el riesgo de los productos
fabricados y respetando el marco normativo armonizado en el MERCOSUR.
4. PROCEDIMIENTOS
4.1 Las inspecciones a los establecimientos fabricantes de productos
médicos y productos para diagnóstico de uso in vitro ubicados en los
Estados Partes deberán ser realizadas por equipos integrados con
inspectores entrenados conforme al programa de capacitación conjunta
aprobado.
4.2 En la realización de las inspecciones deberán ser observados los
siguientes procedimientos:
a) la inspección será realizada por el
Estado Parte Sede (EPS), que deberá elaborar el Acta/Informe de
Inspección que contenga mínimamente las informaciones definidas en el
modelo que consta en el Apéndice de este Anexo;
b) el EPS deberá tomar las medidas apropiadas, de conformidad con los
resultados de la inspección realizada;
c) en el caso que fuese solicitado, el EPS remitirá el Acta/Informe de
Inspección para la consideración del Estado Parte Receptor (EPR)
solicitante;
d) el EPR podrá solicitar informaciones complementarias sobre el
Acta/Informe de Inspección al EPS, si lo considera necesario;
e) el EPR concederá la Certificación de BPF en base al Acta/Informe de
Inspección emitido por el EPS, una vez que todas las informaciones
necesarias para la verificación del cumplimiento de BPF sean cumplidas
y que la conclusión sea satisfactoria;
f) cuando las informaciones presentadas
no fuesen suficientes, la situación podrá ser resuelta mediante la
inspección conjunta al establecimiento, que deberá ser programada entre
los Estados Partes involucrados.
4.3 La autoridad competente del EPS tendrá treinta (30) días corridos
para enviar su respuesta, contados a partir de la fecha de la recepción
de la solicitud por la autoridad competente del EPR, pudiendo:
a) realizar el envío de un Acta/Informe
de Inspección o las informaciones complementarias solicitadas;
b) informar sobre la necesidad de extensión de plazo para el envío de
la documentación solicitada, cuando la empresa esté en el proceso de
cumplimiento de las no conformidades observadas durante la inspección
realizada por la autoridad competente del EPS;
c) informar sobre la imposibilidad de envío de la documentación
solicitada, cuando no haya un Acta/Informe de Inspección válido para la
empresa fabricante o cuando la empresa no se encuentre en condiciones
de exportar productos.
4.4 Para dar cumplimiento a la presente Resolución serán consideradas
válidas aquellas Actas/Informes de Inspecciones realizadas por los EPS
dentro del período de veinticuatro (24) meses anteriores a la fecha de
solicitud de la documentación por el EPR. Los veinticuatro (24) meses
serán contados a partir de la fecha de finalización de la inspección
realizada por la autoridad competente del EPS.
En caso de envío de Actas/Informes de inspecciones realizadas en un
período superior a los mencionados veinticuatro (24) meses, se deberá
adjuntar informe técnico con base en el análisis de riesgo que
justifique la inexistencia de documentos más actuales.
Las Actas/Informes enviadas por el EPS deben corresponder a documentos
de la última inspección realizada en la empresa. Los informes técnicos
y las actas presentadas por los EPS quedan sujetos, en todos los casos,
a la evaluación por parte de las autoridades sanitarias de los EPR.
(Punto sustituido por art. 1° de la Resolución N° 34/2023 del Grupo Mercado Común B.O. 30/12/2024)
4.5 A partir del análisis de los reportes de tecnovigilancia de los
Estados Partes, en función del riesgo potencial de daño para la salud
pública, se podrán realizar inspecciones conjuntas entre los Estados
Partes involucrados.
4.6. La autoridad competente del EPR se compromete a informar a la
autoridad competente del EPS cuando el resultado del intercambio de
documentos previsto en la presente Resolución no permita la
certificación de una empresa. Esta comunicación será realizada en el
menor plazo posible, preferentemente en el momento de notificación a la
empresa interesada del EPR. Para todos los casos la autoridad
competente del EPR deberá informar al EPS sobre su decisión relativa a
la certificación del establecimiento dentro de los treinta (30) días
corridos contados a partir de la fecha en que se tomó la decisión.
5. DISPOSICIONES FINALES
5.1 Las autoridades competentes del EPS deberán informar, con la debida
fundamentación, cualquier modificación en el estado de la certificación
de los establecimientos que exportan a los demás Estados Partes dentro
de los treinta (30) días corridos a partir de la fecha en que dicha
modificación fuera observada.
5.2. El intercambio de documentos previsto en la presente Resolución
deberá ser realizado exclusivamente por los canales oficiales,
acordados entre las autoridades competentes, y deberá mantener el
carácter confidencial de las informaciones técnicas intercambiadas
entre el EPS y el EPR.
Solamente serán considerados válidos para el análisis aquellos
documentos enviados y recibidos por las autoridades de los Estados
Partes involucrados en el proceso de intercambio de informaciones.
5.3. Otras situaciones relacionadas al control y la fiscalización
sanitaria no previstas en esta norma deben ser objeto de tratamiento
específico, mediante negociaciones entre las autoridades competentes de
los Estados Partes involucrados.
Apéndice
ACTA/INFORME DE INSPECCIÓN
EMPRESA SOLICITANTE:
DIRECCIÓN:
PERÍODO DE INSPECCIÓN:
(LUGAR), ____ DE __________ DEL 20__
1. IDENTIFICACIÓN DE LA EMPRESA
SOLICITANTE
1.1 Nombre:
1.2 Dirección:
1.3 Autorización de Funcionamiento N°:
2. INSPECCIÓN
2.1 Período: __/__/__ al __/__/__
2.2 Objetivo de la inspección: verificación del cumplimiento de las
Buenas Prácticas de Fabricación de Productos Médicos y/o Productos para
Diagnóstico de Uso
In Vitro,
conforme a la legislación vigente.
2.3 Tipo de Inspección:
( ) Inicial
( ) Re-inspección
2.4 Fecha de la última inspección: __/__/__
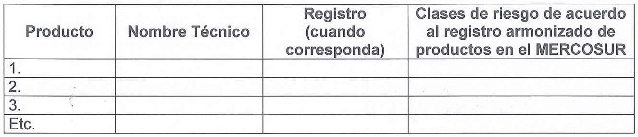
2.5 Listado de los productos fabricados:
Observaciones:
1. Deberán listarse todos los productos fabricados por la planta
inspeccionada, incluyendo los productos en desarrollo y que la empresa
tiene la intención de registrar.
2. Si el listado de productos fuera muy amplio, deberá agregarse como
anexo.
3. PERSONAS CONTACTADAS EN LA EMPRESA
3.1 Personas contactadas en la empresa durante la inspección:
Nombre:
Cargo:
Teléfono: Fax:
Correo electrónico:
Nombre:
Cargo:
Teléfono: Fax:
Correo electrónico:
Nombre:
Cargo:
Teléfono: Fax:
Correo electrónico:
4. LISTA DE PROVEEDORES DE SERVICIOS
Observaciones:
1. Se deberán listar a los proveedores de servicios que pueden influir
en la calidad de los productos fabricados.
2. Si la lista de proveedores que se describe es muy extensa, deberá
agregarse como anexo.
5. INFORMACIONES GENERALES
Describir a la empresa de manera general, el número de empleados, área
de edificación, números de edificios, las características del sitio,
informaciones legales, grupo empresarial y otros datos que el inspector
considere necesarios.
6. REQUISITOS GENERALES DEL SISTEMA DE
CALIDAD
Describir las evidencias relacionadas con los requisitos de la
Responsabilidad Gerencial, Manual de Calidad, Personal, Gestión de
Riesgo y Control de Compras.
6.1 Observación(es):
6.2 No Conformidad(es):
7. DOCUMENTOS Y REGISTROS DE CALIDAD
Describir procedimientos relacionados con control de documentos y
registros.
7.1 Observación(es):
7.2 No Conformidad(es):
8. CONTROL DE DISEÑO Y REGISTRO
MAESTRO DE PRODUCTO
Describir las evidencias relativas a los requisitos de control de
diseño, registro histórico de diseño y registro maestro de producto.
8.1 Observación(es):
8.2 No Conformidad(es):
9. CONTROLES DE PROCESO Y PRODUCCIÓN
Describir los aspectos relacionados con las instalaciones de la
empresa, controles ambientales, de salud ocupacional, procedimientos y
evidencias relacionadas con los controles de las diferentes etapas de
la producción, envasado y etiquetado de los productos, liberación de
los productos, programas de mantenimiento, inspección y ensayos,
calibración, validación y control de cambios.
9.1 Observación(es):
9.2 No Conformidad(es):
10. MANIPULACIÓN, ALMACENAMIENTO,
DISTRIBUCIÓN Y TRAZABILIDAD
Describir procedimientos, registros y evidencias que se relacionen con
los requisitos de manipulación, almacenamiento, identificación,
trazabilidad de los componentes y productos terminados, la distribución
de los productos terminados, y los procedimientos para los componentes
y productos no conformes.
10.1 Observación(es):
10.2 No Conformidad(es):
11. ACCIONES CORRECTIVAS Y PREVENTIVAS
Describir los procedimientos y registros de acciones correctivas y
preventivas, acciones de campo y retiro de productos, gestión de
reclamos y auditorías de calidad.
11.1 Observación(es):
11.2 No Conformidad(es):
12. INSTALACIÓN Y ASISTENCIA TÉCNICA
Describir los procedimientos y registros relacionados a la instalación
y la asistencia técnica de los productos.
12.1 Observación(es):
12.2 No Conformidad(es):
13. TÉCNICAS ESTADÍSTICAS
Describir los procedimientos relacionados con las técnicas estadísticas
adoptadas para la evaluación del desempeño del sistema de calidad y
capacidad del proceso para atender las especificaciones establecidas,
así como para definir los planes de muestreo.
13.1 Observación(es):
13.2 No Conformidad(es):
14. CONSIDERACIONES GENERALES /
EVALUACIÓN DEL RIESGO / RECOMENDACIONES
Registrar las consideraciones generales y las recomendaciones
formuladas a la empresa por el equipo de inspectores. En el caso de una
infracción sanitaria, las medidas adoptadas por el equipo de
inspectores deben ser registradas y las copias de los documentos
pertinentes se deberán adjuntar al Acta/Informe de la Inspección.
15. CONCLUSIÓN
(_) SATISFACTORIA
(_) CON OBSERVACIONES Y/O NO CONFORMIDADES
Plazo de cumplimiento contado a partir de la recepción del Acta/Informe:
(_) INSATISFACTORIA
16. EQUIPO DE INSPECTORES
IF-2024-07852970-APN-DRI#ANMAT