ADMINISTRACIÓN NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGÍA MÉDICA
Disposición 6559/2025
DI-2025-6559-APN-ANMAT#MS
Ciudad de Buenos Aires, 05/09/2025
VISTO el expediente n° EX-2025-97726545-APN-DERM#ANMAT, la Ley Nº
16.463, el Decreto N° 150 del 20 de enero de 1992 (T.O.1993) y las
Disposiciones ANMAT Nros. 3185 del 25 de junio de 1999, 5040 del 6 de
septiembre de 2006 y sus modificatorias, 556 del 5 de febrero de 2009,
758 del 23 de febrero de 2009 y sus modificatoria, 6677 del 1 de
noviembre de 2010 y su modificatoria, 4133 del 19 de julio de 2012,
4326 del 26 de julio de 2012, 1918 del 11 de abril de 2013, 5640 del 14
de julio de 2022 y su modificatoria, y
CONSIDERANDO:
Que por la Disposición Nº 3185/99 de esta Administración Nacional de
Medicamentos, Alimentos y Tecnología Médica (ANMAT) se establecieron
las exigencias de estudios de biodisponibilidad/bioequivalencia entre
productos, y se adoptó el criterio para su implementación gradual de
acuerdo al riesgo sanitario de su ingrediente farmacéutico activo (IFA).
Que la aplicación efectiva de la referida Disposición ANMAT Nº 3185/99
ha permitido el desarrollo, ejecución y evaluación de los resultados de
los estudios de Bioequivalencia entre medicamentos, que las empresas
titulares de certificados de especialidades medicinales han presentado
ante esta Administración.
Que el advenimiento de la Clasificación Biofarmacéutica (CBF) ha resultado un adelanto en el entorno de la bioequivalencia.
Que la mencionada clasificación se basa en la solubilidad y
permeabilidad del ingrediente farmacéutico activo y en el tiempo de
disolución de la forma farmacéutica sólida oral.
Que esta ANMAT, mediante la Disposición Nº 758/09, adoptó los criterios
de dicha clasificación, estableciendo que sólo los IFAs pertenecientes
a las categorías II (baja solubilidad - alta permeabilidad) y IV (baja
solubilidad - baja permeabilidad) requieren la realización de estudios
de bioequivalencia.
Que la misma Disposición estableció las condiciones por las cuales las
formulaciones proporcionalmente similares al producto multifuente que
ha probado ser bioequivalente al producto comparador de referencia
pueden demostrar su equivalencia mediante estudios in vitro.
Que, asimismo, cabe destacar que por la Disposición ANMAT Nº 556/09 se
aprobó la guía para aplicar en los cambios de escala y cambios
posteriores al registro de medicamentos sujetos a demostración de
bioequivalencia.
Que en razón de sus características farmacocinéticas deviene necesario
exigir estudios de bioequivalencia a las especialidades medicinales que
contengan como IFA los hipoglucemiantes orales categorías II y IV
(según la Clasificación Biofarmacéutica).
Que la Dirección de Investigación Clínica y Gestión del Registro de
Medicamentos del Instituto Nacional de Medicamentos y la Dirección de
Asuntos Jurídicos han tomado intervención en el ámbito de su
competencia.
Que se actúa en virtud de las facultades conferidas por el Decreto N° 1490/92 y sus modificatorios.
Por ello,
LA ADMINISTRADORA NACIONAL DE LA ADMINISTRACIÓN NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGÍA MÉDICA
DISPONE:
ARTÍCULO 1º.- Incorpóranse a las exigencias de realización de estudios
de bioequivalencia/biodisponibilidad, establecidas por la Disposición
ANMAT Nº 3185/99, a los ingredientes farmacéuticos activos que figuran
en el Anexo I (IF-2025-98493658-APN-DERM#ANMAT) que forma parte de la
presente disposición. Los respectivos Productos de Referencia se listan
en el ANEXO II (IF-2025-98490243-APN-DERM#ANMAT) que forma parte de la
presente disposición.
ARTÍCULO 2º.-Los resultados de los estudios de bioequivalencia de las
especialidades medicinales alcanzadas por el artículo 1°, que respondan
a los criterios de aceptación establecidos en la normativa vigente,
deberán ser presentados en un plazo no mayor de ciento ochenta (180)
días corridos a partir de la entrada en vigencia de la presente.
Vencido el referido plazo, si no se efectuó dicha presentación o si los
resultados no demostraron bioequivalencia con el producto de
referencia, se procederá, sin necesidad de intimación previa, a la
suspensión de la comercialización de las especialidades medicinales
involucradas, salvo que consideraciones de salud pública así lo
ameriten.
ARTÍCULO 3º.- Los laboratorios titulares de especialidades medicinales
que contengan alguno de los ingredientes farmacéuticos activos
mencionados en el Anexo I de la presente disposición, con formulaciones
proporcionalmente similares al producto multifuente que demostró ser
bioequivalente al producto de referencia, podrán demostrar su
equivalencia mediante estudios in vitro.
ARTÍCULO 4º.- El incumplimiento de las obligaciones establecidas en la
presente disposición hará pasible a los infractores de las sanciones
previstas en la Ley Nº 16.463 y el Decreto Nº 341/92, sin perjuicio de
las demás acciones que pudieran corresponder.
ARTÍCULO 5°.- La presente disposición entrará en vigencia el día de su publicación en el Boletín Oficial.
ARTÍCULO 6°.- Dese a la Dirección Nacional del Registro Oficial para su
publicación. Dese al Instituto Nacional de Medicamentos, a la Dirección
de Investigación Clínica y Gestión del Registro de Medicamentos, a la
Dirección de Gestión de Información Técnica y a la Dirección de
Relaciones Institucionales. Comuníquese a las Cámaras de Especialidades
Medicinales (CILFA, CAEMe, COOPERALA, CAPGEN, CAPEMVeL, SAFYBI),
Confederación Médica de la República Argentina (COMRA) y la
Confederación Farmacéutica Argentina (COFA), y demás entidades
representativas del sector. Cumplido, archívese.
Nelida Agustina Bisio
NOTA: El/los Anexo/s que integra/n este(a) Disposición se publican en la edición web del BORA -www.boletinoficial.gob.ar-
e. 08/09/2025 N° 65025/25 v. 08/09/2025
(Nota
Infoleg:
Los anexos referenciados en la presente norma han sido extraídos de la
edición web de Boletín Oficial)
ANEXO I
INGREDIENTES FARMACÉUTICOS ACTIVOS QUE DEBEN REALIZAR ESTUDIOS DE
BIOEQUIVALENCIA DE ACUERDO A LO ESTABLECIDO EN LAS DISPOSICIONES
(ANMAT) Nº 5040/06 y 1746/07 y 5640/22
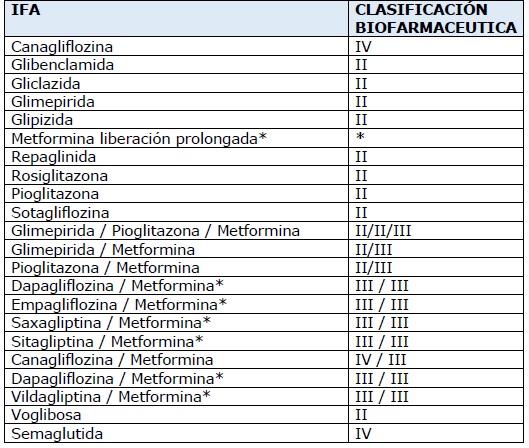
*Si bien la Metformina, de acuerdo a
la Clasificación Biofarmacéutica, es de categoría III se la considera
como de alta solubilidad con absorción limitada (saturable e
incompleta) y cuya farmacocinética no es lineal. Por lo tanto, requiere
estudios de bioequivalencia.
Ref:
https://www.ema.europa.eu/en/documents/scientificguideline/metformin-immediate-release-film-coated-tablets-500-850-
1000-mg-1000-mg-5ml-oral-solution-product-specificbioequivalence- guidance-revision-1_en.pdf
IF-2025-98493658-APN-DERM#ANMAT

ANEXO II
LISTADO DE INGREDIENTES FARMACÉUTICOS ACTIVOS DEL GRUPO DE HIPOGLUCEMIANTES ORALES DE REFERENCIA
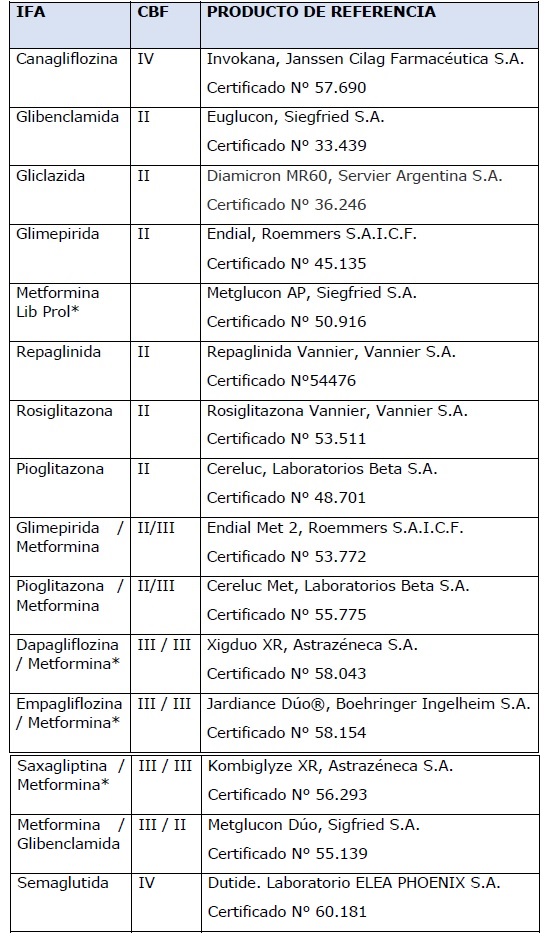
*Si bien la Metformina, de acuerdo a
la Clasificación Biofarmacéutica, es de categoría III se la considera
como de alta solubilidad con absorción limitada (saturable e
incompleta) y cuya farmacocinética no es lineal. Por lo tanto, requiere
estudios de bioequivalencia.
Ref:https://www.ema.europa.eu/en/documents/scientificguideline/metformin-immediate-release-film-coated-tablets-500-850-
1000-mg-1000-mg-5ml-oral-solution-product-specificbioequivalence-
guidance-revision-1_en.pdf
IF-2025-98490243-APN-DERM#ANMAT

